

As well as also for Metal & Stone Cutting Free Activators - necessary
Flux (metallurgy)
Chemical used in metallurgy for cleaning or purifying molten metal


In metallurgy, a flux (from Latin fluxus 'flow') is a chemical cleaning agent, flowing agent, or purifying agent. Fluxes may have more than one function at a time. They are used in both extractive metallurgy and metal joining.
Some of the earliest known fluxes were sodium carbonate, potash, charcoal, coke, borax,[1]lime,[2]lead sulfide[3] and certain minerals containing phosphorus. Iron ore was also used as a flux in the smelting of copper. These agents served various functions, the simplest being a reducing agent, which prevented oxides from forming on the surface of the molten metal, while others absorbed impurities into the slag, which could be scraped off the molten metal.[4]
Fluxes are also used in foundries for removing impurities from molten nonferrous metals such as aluminium, or for adding desirable trace elements such as titanium.
As cleaning agents, fluxes facilitate soldering, brazing, and welding by removing oxidation from the metals to be joined. In some applications molten flux also serves as a heat-transfer medium, facilitating heating of the joint by the soldering tool or molten solder.
Uses[edit]
Metal joining[edit]
In high-temperature metal joining processes (welding, brazing and soldering), flux is a substance that is nearly inert at room temperature, but which becomes strongly reducing at elevated temperatures, preventing oxidation of the base and filler materials. The role of flux is typically dual: dissolving the oxides already present on the metal surface, which facilitates wetting by molten metal, and acting as an oxygen barrier by coating the hot surface, preventing its oxidation.
For example, tin-lead solder[5] attaches very well to copper, but poorly to the various oxides of copper, which form quickly at soldering temperatures. By preventing the formation of metal oxides, flux enables the solder to adhere to the clean metal surface, rather than forming beads, as it would on an oxidized surface.
Soldering[edit]
In soldering of metals, flux serves a threefold purpose: it removes any oxidized metal from the surfaces to be soldered, seals out air thus preventing further oxidation, and by facilitating amalgamation improves wetting characteristics of the liquid solder.[6] Some fluxes are corrosive, so the parts have to be cleaned with a damp sponge or other absorbent material after soldering to prevent damage. Several types of flux are used in electronics.[7]
A number of standards exist to define the various flux types. The principal standard is J-STD-004.
Various tests, including the ROSE test, may be used after soldering to check for the presence of ionic or other contaminants that could cause short circuits or other problems.
Brazing and silver soldering[edit]
Brazing (sometimes known as silver soldering or hard soldering) requires a much higher temperature than soft soldering, sometimes over 850 °C. As well as removing existing oxides, rapid oxidation of the metal at the elevated temperatures has to be avoided. This means that fluxes need to be more aggressive and to provide a physical barrier.[8] Traditionally borax was used as a flux for brazing, but there are now many different fluxes available, often using active chemicals such as fluorides[9] as well as wetting agents. Many of these chemicals are toxic and due care should be taken during their use.
Smelting[edit]
Main article: Smelting § Fluxes
In the process of smelting, inorganic chlorides, fluorides (see fluorite), limestone and other materials are designated as "fluxes" when added to the contents of a smelting furnace or a cupola for the purpose of purging the metal of chemical impurities such as phosphorus, and of rendering slag more liquid at the smelting temperature. The slag is a liquid mixture of ash, flux, and other impurities. This reduction of slag viscosity with temperature, increasing the flow of slag in smelting, is the origin of the word flux in metallurgy.
The flux most commonly used in iron and steel furnaces is limestone, which is charged in the proper proportions with the iron and fuel.
Drawbacks[edit]
Fluxes have several serious drawbacks:
- Corrosivity, which is mostly due to the aggressive compounds of the activators; hygroscopic properties of the flux residues may aggravate the effects
- Interference with test equipment, which is due to the insulating residues deposited on the test contacts on electronic circuit boards
- Interference with machine vision systems when the layer of flux or its remains is too thick or improperly located
- Contamination of sensitive parts, e.g. facets of laser diodes, contacts of connectors and mechanical switches, and MEMS assemblies
- Deterioration of electrical properties of printed circuit boards, as soldering temperatures are above the glass transition temperature of the board material and flux components (e.g. glycols, or chloride and bromide ions) can diffuse into its matrix; e.g. water-soluble fluxes containing polyethylene glycol were demonstrated to have such impact[10]
- Deterioration of high-frequency circuit performance by flux residues
- Deterioration of surface insulation resistance, which tends to be as much as three orders of magnitude lower than the bulk resistance of the material
- Electromigration and growth of whiskers between nearby traces, aided by ionic residues, surface moisture and a bias voltage
- The fumes liberated during soldering have adverse health effects, and volatile organic compounds can be outgassed during processing
- The solvents required for post-soldering cleaning of the boards are expensive and may have adverse environmental impact
In special cases the drawbacks are sufficiently serious to warrant using fluxless techniques.
Dangers[edit]
Acid flux types (not used in electronics) may contain hydrochloric acid, zinc chloride or ammonium chloride, which are harmful to humans. Therefore, flux should be handled with gloves and goggles, and used with adequate ventilation.
Prolonged exposure to rosin fumes released during soldering can cause occupational asthma (formerly called colophony disease[11] in this context) in sensitive individuals, although it is not known which component of the fumes causes the problem.[12]
While molten solder has low tendency to adhere to organic materials, molten fluxes, especially of the resin/rosin type, adhere well to fingers. A mass of hot sticky flux can transfer more heat to skin and cause more serious burns than a comparable particle of non-adhering molten metal, which can be quickly shaken off. In this regard, molten flux is similar to molten hot glue.
Fluxless techniques[edit]
In some cases the presence of flux is undesirable; flux traces interfere with e.g. precision optics or MEMS assemblies. Flux residues also tend to outgas in vacuum and space applications, and traces of water, ions and organic compounds may adversely affect long-term reliability of non-hermetic packages. Trapped flux residues are also the cause of most voids in the joints. Flux-less techniques are therefore desirable there.[13]
For successful soldering and brazing, the oxide layer has to be removed from both the surfaces of the materials and the surface of the filler metal preform; the exposed surfaces also have to be protected against oxidation during heating. Flux-coated preforms can also be used to eliminate flux residue entirely from the soldering process.[14]
Protection of the surfaces against further oxidation is relatively simple, by using vacuum or inert atmosphere. Removal of the native oxide layer is more troublesome; physical or chemical cleaning methods have to be employed and the surfaces can be protected by e.g. gold plating. The gold layer has to be sufficiently thick and non-porous to provide protection for reasonable storage time. Thick gold metallization also limits choice of soldering alloys, as tin-based solders dissolve gold and form brittle intermetallics, embrittling the joint. Thicker gold coatings are usually limited to use with indium-based solders and solders with high gold content.[citation needed]
Removal of the oxides from the solder preform is also troublesome. Fortunately some alloys are able to dissolve the surface oxides in their bulk when superheated by several degrees above their melting point; the Sn-Cu1 and Sn-Ag4 require superheating by 18–19 °C, the Sn-Sb5 requires as little as 10 °C, but the Sn-Pb37 alloy requires 77 °C above its melting point to dissolve its surface oxide.[citation needed] The self-dissolved oxide degrades the solder's properties and increases its viscosity in molten state, however, so this approach is not optimal.
Solder preforms are preferred to be with high volume-to-surface ratio, as that limits the amount of oxide being formed. Pastes have to contain smooth spherical particles, preforms are ideally made of round wire. The problem with preforms[which?] can be also sidestepped by depositing the solder alloy directly on the surfaces of the parts or substrates, by chemical or electrochemical means for example.[citation needed]
A protective atmosphere with chemically reducing properties can be beneficial in some cases. Molecular hydrogen can be used to reduce surface oxides of tin and indium at temperatures above 430 and 470 °C; for zinc the temperature is above 500 °C, where zinc is already becoming volatilized. (At lower temperatures the reaction speed is too slow for practical applications.) Very low partial pressures of oxygen and water vapor have to be achieved for the reaction to proceed.[citation needed]
Other reactive atmospheres are also in use. Vapors of formic acid and acetic acid are the most commonly used. Carbon monoxide and halogen gases (for example carbon tetrafluoride, sulfur hexafluoride, or dichlorodifluoromethane) require fairly high temperatures for several minutes to be effective.[citation needed]
Atomic hydrogen is much more reactive than molecular hydrogen. In contact with surface oxides it forms hydroxides, water, or hydrogenated complexes, which are volatile at soldering temperatures. The most practical dissociation method is probably an electrical discharge.[ambiguous] Argon-hydrogen gas compositions with hydrogen concentration below the low flammable limit can be used, eliminating the safety issues. The operation has to be performed at low pressure, as the stability of atomic hydrogen at atmospheric pressure is insufficient. Such hydrogen plasma can be used for fluxless reflow soldering.[citation needed]
Active atmospheres are relatively common in furnace brazing; due to the high process temperatures the reactions are reasonably fast. The active ingredients are usually carbon monoxide (possibly in the form of combusted fuel gas) and hydrogen. Thermal dissociation of ammonia yields an inexpensive mixture of hydrogen and nitrogen.[citation needed]
Bombardment with atomic particle beams can remove surface layers at a rate of tens of nanometers per minute. The addition of hydrogen to the plasma[which?] augments the removal efficiency by chemical mechanisms.[citation needed]
Mechanical agitation is another possibility for disrupting the oxide layer. Ultrasound can be used for assisting tinning and soldering; an ultrasonic transducer can be mounted on the soldering iron, in a solder bath, or in the wave for wave soldering. The oxide disruption and removal involves cavitation effects between the molten solder and the base metal surface. A common application of ultrasound fluxing is in tinning of passive parts (active parts do not cope well with the mechanical stresses involved); even aluminium can be tinned this way. The parts can then be soldered or brazed conventionally.[citation needed]
Mechanical rubbing of a heated surface with molten solder can be used for coating the surface. Both surfaces to be joined can be prepared this way, then placed together and reheated. This technique was formerly used to repair small damages on aluminium aircraft skins.[citation needed]
A very thin layer of zinc can be used for joining aluminium parts. The parts have to be perfectly machined, or pressed together, due to the small volume of filler metal. At high temperature applied for long time, the zinc diffuses away from the joint. The resulting joint does not present a mechanical weakness and is corrosion-resistant. The technique is known as diffusion soldering.[citation needed]
Fluxless brazing of copper alloys can be done with self-fluxing filler metals. Such metals contain an element capable of reaction with oxygen, usually phosphorus. A good example is the family of copper-phosphorus alloys.[citation needed]
Properties[edit]
Fluxes have several important properties:
- Activity – the ability to dissolve existing oxides on the metal surface and promote wetting with solder. Highly active fluxes are often acidic or corrosive in nature.
- Corrosivity – the promotion of corrosion by the flux and its residues. Most active fluxes tend to be corrosive at room temperatures and require careful removal. As activity and corrosivity are linked, the preparation of surfaces to be joined should allow use of milder fluxes. Some water-soluble flux residues are hygroscopic, which causes problems with electrical resistance and contributes to corrosion. Fluxes containing halides and mineral acids are highly corrosive and require thorough removal. Some fluxes, especially those based on borax used for brazing, form very hard glass-like coatings that are difficult to remove.
- Cleanability – the difficulty of removal of flux and its residues after the soldering operation. Fluxes with higher content of solids tend to leave larger amount of residues; thermal decomposition of some vehicles also leads to formation of difficult-to-clean, polymerized and possibly even charred deposits (a problem especially for hand soldering). Some flux residues are soluble in organic solvents, others in water, some in both. Some fluxes are no-clean, as they are sufficiently volatile or undergo thermal decomposition to volatile products, that they do not require the cleaning step. Other fluxes leave non-corrosive residues that can be left in place. However, flux residues can interfere with subsequent operations; they can impair adhesion of conformal coatings, or act as undesired insulation on connectors and contact pads for test equipment.
- Residue tack – the stickiness of the surface of the flux residue. When not removed, the flux residue should have smooth, hard surface. Tacky surfaces tend to accumulate dust and particulates, which causes issues with electrical resistance; the particles themselves can be conductive or they can be hygroscopic or corrosive.
- Volatility – this property has to be balanced to facilitate easy removal of solvents during the preheating phase but to not require too frequent replenishing of solvent in the process equipment.
- Viscosity – especially important for solder pastes, which have to be easy to apply but also thick enough to stay in place without spreading to undesired locations. Solder pastes may also function as a temporary adhesive for keeping electronic parts in place before and during soldering. Fluxes applied by e.g. foam require low viscosity.
- Flammability – relevant especially for glycol-based vehicles and for organic solvents. Flux vapors tend to have low autoignition temperature and present a risk of a flash fire when the flux comes in contact with a hot surface.
- Solids – the percentage of solid material in the flux. Fluxes with low solids, sometimes as little as 1–2%, are called low solids flux, low-residue flux, or no clean flux. They are often composed of weak organic acids, with addition of small amount of rosin or other resins.
- Conductivity – some fluxes remain conductive after soldering if not cleaned properly, leading to random malfunctions on circuits with high impedances. Different types of fluxes are differently prone to cause these issues.
Composition[edit]
Fluxes for metal joining[edit]
The composition of fluxes is tailored for the required properties - the base metals and their surface preparation (which determine the composition and thickness of surface oxides), the solder (which determines the wetting properties and the soldering temperature), the corrosion resistance and ease of removal, and others.
Fluxes for soft soldering are typically of organic nature, though inorganic fluxes, usually based on halogenides or acids, are also used in non-electronics applications. Fluxes for brazing operate at significantly higher temperatures and are therefore mostly inorganic; the organic compounds tend to be of supplementary nature, e.g. to make the flux sticky at low temperature so it can be easily applied.
The surface of the tin-based solder is coated predominantly with tin oxides; even in alloys the surface layer tends to become relatively enriched by tin. Fluxes for indium and zinc based solders have different compositions than fluxes for ordinary tin-lead and tin-based solders, due to different soldering temperatures and different chemistry of the oxides involved.
Organic fluxes are unsuitable for flame soldering and flame brazing, as they tend to char and impair solder flow.
Some metals are classified as "unsolderable" in air, and have to be either coated with another metal before soldering or special fluxes or protective atmospheres have to be used. Such metals are beryllium, chromium, magnesium, titanium, and some aluminium alloys.
Fluxes for high-temperature soldering differ from the fluxes for use at lower temperatures. At higher temperatures even relatively mild chemicals have sufficient oxide-disrupting activity, but the metal oxidation rates become fairly high; the barrier function of the vehicle therefore becomes more important than the fluxing activity. High molecular weight hydrocarbons are often used for this application; a diluent with a lower molecular weight, boiling off during the preheat phase, is usually used to aid application.[15]
Common fluxes are ammonium chloride or resin acids (contained in rosin) for soldering copper and tin; hydrochloric acid and zinc chloride for soldering galvanizediron (and other zinc surfaces); and borax for brazing, braze-welding ferrous metals, and forge welding.
Organic fluxes[edit]
Organic fluxes typically consist of four major components:[16]
- Activators – chemicals disrupting/dissolving the metal oxides. Their role is to expose unoxidized, easily wettable metal surface and aid soldering by other means, e.g. by exchange reactions with the base metals.
- Vehicles – high-temperature tolerant chemicals in the form of non-volatile liquids or solids with suitable melting point; they are generally liquid at soldering temperatures. Their role is to act as an oxygen barrier to protect the hot metal surface against oxidation, to dissolve the reaction products of activators and oxides and carry them away from the metal surface, and to facilitate heat transfer. Solid vehicles tend to be based on natural or modified rosin (mostly abietic acid, pimaric acid, and other resin acids) or natural or synthetic resins. Water-soluble organic fluxes tend to contain vehicles based on high-boiling polyols - glycols, diethylene glycol and higher polyglycols, polyglycol-based surfactants and glycerol.
- Solvents – added to facilitate processing and deposition to the joint. Solvents are typically dried out during preheating before the soldering operation; incomplete solvent removal may lead to boiling off and spattering of solder paste particles or molten solder.
- Additives – numerous other chemicals modifying the flux properties. Additives can be surfactants (especially nonionic), corrosion inhibitors, stabilizers and antioxidants, tackifiers, thickeners and other rheological modifiers (especially for solder pastes), plasticizers (especially for flux-cored solders), and dyes.
Inorganic fluxes[edit]
Inorganic fluxes contain components playing the same role as in organic fluxes. They are more often used in brazing and other high-temperature applications, where organic fluxes have insufficient thermal stability. The chemicals used often simultaneously act as both vehicles and activators; typical examples are borax, borates, fluoroborates, fluorides and chlorides. Halogenides are active at lower temperatures than borates, and are therefore used for brazing of aluminium and magnesium alloys; they are however highly corrosive.
Behavior of activators[edit]
The role of the activators is primarily disruption and removal of the oxide layer on the metal surface (and also the molten solder), to facilitate direct contact between the molten solder and metal. The reaction product is usually soluble or at least dispersible in the molten vehicle. The activators are usually either acids, or compounds that release acids at elevated temperature.
The general reaction of oxide removal is:
- Metal oxide + Acid → Salt + Water
Salts are ionic in nature and can cause problems from metallic leaching or dendrite growth, with possible product failure. In some cases, particularly in high-reliability applications, flux residues must be removed.
The activity of the activator generally increases with temperature, up to a certain value where activity ceases, either due to thermal decomposition or excessive volatilization. However the oxidation rate of the metals also increases with temperature.
At high temperatures, copper oxide reacts with hydrogen chloride to water-soluble and mechanically weak copper chloride, and with rosin to salts of copper and abietic acid which is soluble in molten rosin.
Some activators may also contain metal ions, capable of exchange reaction with the underlying metal; such fluxes aid soldering by chemically depositing a thin layer of easier solderable metal on the exposed base metal. An example is the group of fluxes containing zinc, tin or cadmium compounds, usually chlorides, sometimes fluorides or fluoroborates.
Inorganic activators[edit]
Common high-activity activators are mineral acids, often together with halides, amines, water or alcohols:
Inorganic acids are highly corrosive to metals even at room temperature, which causes issues during storage, handling and applications. As soldering involves high temperatures, compounds that decompose or react, with acids as products, are frequently used:
Rosin fluxes[edit]

The terms resin flux and rosin flux are ambiguous and somewhat interchangeable, with different vendors using different assignments. Generally, fluxes are labeled as rosin if the vehicle they are based on is primarily natural rosin. Some manufactures reserve "rosin" designation for military fluxes based on rosin (R, RMA and RA compositions) and label others as "resin".
Rosin has good flux properties. A mixture of organic acids (resin acids, predominantly abietic acid, with pimaric acid, isopimaric acid, neoabietic acid, dihydroabietic acid, and dehydroabietic acid), rosin is a glassy solid, virtually nonreactive and noncorrosive at normal temperature, but liquid, ionic and mildly reactive to metal oxides at molten state. Rosin tends to soften between 60–70 °C and is fully fluid at around 120 °C; molten rosin is weakly acidic and is able to dissolve thinner layers of surface oxides from copper without further additives. For heavier surface contamination or improved process speed, additional activators can be added.
There are several possible activator groups for rosins:
There are three types of rosin: gum rosin (from pine tree oleoresin), wood rosin (obtained by extraction of tree stumps), and tall oil rosin (obtained from tall oil, a byproduct of kraft paper process). Gum rosin has a milder odor and lower tendency to crystallize from solutions than wood rosin, and is therefore preferred for flux applications. Tall oil rosin finds increased use due to its higher thermal stability and therefore lower tendency to form insoluble thermal decomposition residues. The composition and quality of rosin differs by the tree type, and also by location and even by year. In Europe, rosin for fluxes is usually obtained from a specific type of Portuguese pine, in America a North Carolina variant is used.[17]
Natural rosin can be used as-is, or can be chemically modified by e.g. esterification, polymerization, or hydrogenation. The properties being altered are increased thermal stability, better cleanability, altered solution viscosity, and harder residue (or conversely, softer and more tacky residue). Rosin can be also converted to a water-soluble rosin flux, by formation of an ethoxylated rosin amine, an adduct with a polyglycol and an amine.
One of the early fluxes was a mixture of equal amounts of rosin and vaseline. A more aggressive early composition was a mixture of saturated solution of zinc chloride, alcohol, and glycerol.[18]
Fluxes can be also prepared from synthetic resins, often based on esters of polyols and fatty acids. Such resins have improved fume odor and lower residue tack, but their fluxing activity and solubility tend to be lower than that of natural resins.
Rosin flux grades[edit]
Rosin fluxes are categorized by grades of activity: L for low, M for moderate, and H for high. There are also other abbreviations for different rosin flux grades:[17][19]
- R (Rosin) – pure rosin, no activators, low activity, mildest
- WW (Water-White) – purest rosin grade, no activators, low activity, sometimes synonymous with R
- RMA (Rosin Mildly Activated) - contains mild activators, typically no halides
- RA (Rosin Activated) – rosin with strong activators, high activity, contains halides
- OA (Organic Acid) – rosin activated with organic acids, high activity, highly corrosive, aqueous cleaning
- SA (Synthetically Activated) – rosin with strong synthetic activators, high activity; formulated to be easily soluble in organic solvents (chlorofluorocarbons, alcohols) to facilitate cleaning
- WS (Water-Soluble) – usually based on inorganic or organic halides; highly corrosive residues
- SRA (Superactivated rosin) – rosin with very strong activators, very high activity
- IA (Inorganic Acid) – rosin activated with inorganic acids (usually hydrochloric acid or phosphoric acid), highest activities, highly corrosive
R, WW, and RMA grades are used for joints that can not be easily cleaned or where there is too high corrosion risk. More active grades require thorough cleaning of the residues. Improper cleaning can actually aggravate the corrosion by releasing trapped activators from the flux residues.
Special fluxes[edit]
Fluxes for soldering certain metals[edit]
Some materials are very difficult to solder. In some cases special fluxes have to be employed.
Aluminum and its alloys[edit]
Aluminium and its alloys are difficult to solder due to the formation of the passivation layer of aluminium oxide. The flux has to be able to disrupt this layer and facilitate wetting by solder. Salts or organic complexes of some metals can be used; the salt has to be able to penetrate the cracks in the oxide layer.[citation needed] The metal ions, more noble than aluminium, then undergo a redox reaction, dissolve the surface layer of aluminium and form a deposit there. This intermediate layer of another metal then can be wetted with a solder.
One example of such flux is a composition of triethanolamine, fluoroboric acid, and cadmium fluoroborate. More than 1% magnesium in the alloy impairs the flux action, however, as the magnesium oxide layer is more refractory. Another possibility is an inorganic flux composed of zinc chloride or tin(II) chloride,[20]ammonium chloride, and a fluoride (e.g. sodium fluoride). Presence of silicon in the alloy impairs the flux effectivity, as silicon does not undergo the exchange reaction aluminium does.
Magnesium alloys[edit]
Magnesium alloys. A putative flux for soldering these alloys at low temperature is molten acetamide. Acetamide dissolves surface oxides on both aluminium and magnesium; promising experiments were done with its use as a flux for a tin-indium solder on magnesium.[citation needed]
Stainless steel[edit]
Stainless steel is material which is difficult to solder because of its stable, self-healing surface oxide layer and its low thermal conductivity. A solution of zinc chloride in hydrochloric acid is a common flux for stainless steels; it has however to be thoroughly removed afterwards as it would cause pitting corrosion. Another highly effective flux is phosphoric acid; its tendency to polymerize at higher temperatures however limits its applications.
Metal salts as flux in hot corrosion[edit]
Hot corrosion can affect gas turbines operating in high salt environments (e.g., near the ocean). Salts, including chlorides and sulfates, are ingested by the turbines and deposited in the hot sections of the engine; other elements present in fuels also form salts, e.g. vanadates. The heat from the engine melts these salts which then can flux the passivating oxide layers on the metal components of the engine, allowing corrosion to occur at an accelerated rate.
List of fluxes[edit]
This section needs to be updated. The reason given is: Does not appear to reflect modern ingredients in use, including most mentioned earlier in this article.. Please help update this article to reflect recent events or newly available information.(March 2021) |
Flux recovery[edit]
During the submerged arc welding process, not all flux turns into slag. Depending on the welding process, 50% to 90% of the flux can be reused.[22]
Standards[edit]
Solder fluxes are specified according to several standards.
ISO 9454-1 and DIN EN 29454-1[edit]
The most common standard in Europe is ISO 9454-1 (also known as DIN EN 29454-1).[23]
This standard specifies each flux by a four-character code: flux type, base, activator, and form. The form is often omitted.
| Flux type | Base | Activator | Form |
|---|---|---|---|
| 1 Resin |
| ||
| 2 Organic |
| ||
| 3 Inorganic | |||
Therefore, 1.1.2 means rosin flux with halides.
DIN 8511[edit]
The older German DIN 8511 specification is still often in use in shops. In the table below, note that the correspondence between DIN 8511 and ISO 9454-1 codes is not one-to-one.
| Residues | DIN 8511 | ISO 9454-1 | Description |
|---|---|---|---|
| Strongly corrosive | F-SW-11 | 3.2.2 | Inorganic acid other than phosphoric |
| Strongly corrosive | F-SW-12 | 3.1.1 | Ammonium chloride |
| Strongly corrosive | F-SW-13 | 3.2.1 | Phosphoric acid |
| Weakly corrosive | F-SW-21 | 3.1.1 | Ammonium chloride |
| Weakly corrosive | F-SW-22 | 3.1.2 | Inorganic salts without ammonium chloride |
| Weakly corrosive | F-SW-23 | 2.1.3 | Organic water-soluble without halides |
| Weakly corrosive | F-SW-23 | 2.2.1 | Organic water-insoluble without activators |
| Weakly corrosive | F-SW-23 | 2.2.3 | Organic water-insoluble without halides |
| Weakly corrosive | F-SW-24 | 2.1.1 | Organic water-soluble without activators |
| Weakly corrosive | F-SW-24 | 2.1.3 | Organic water-soluble without halides |
| Weakly corrosive | F-SW-24 | 2.2.3 | Organic water-insoluble without halides |
| Weakly corrosive | F-SW-25 | 2.1.2 | Organic water-soluble with halides |
| Weakly corrosive | F-SW-25 | 2.2.2 | Organic water-insoluble with halides |
| Weakly corrosive | F-SW-26 | 1.1.2 | Rosin with halides |
| Weakly corrosive | F-SW-27 | 1.1.3 | Rosin without halides |
| Weakly corrosive | F-SW-28 | 1.2.2 | Rosin-free resin with halides |
| Non-corrosive | F-SW-31 | 1.1.1 | Rosin without activators |
| Non-corrosive | F-SW-32 | 1.1.3 | Rosin without halides |
| Non-corrosive | F-SW-33 | 1.2.3 | Rosin-free resin without halides |
| Non-corrosive | F-SW-34 | 2.2.3 | Organic water-insoluble without halides |
J-STD-004[edit]
One standard increasingly used (e.g. in the United States) is J-STD-004. It is very similar to DIN EN 61190-1-1.
Four characters (two letters, then one letter, and last a number) represent flux composition, flux activity, and whether activators include halides:[24]
- First two letters: Base
- RO: rosin
- RE: resin
- OR: organic
- IN: inorganic
- Third letter: Activity
- L: low
- M: moderate
- H: high
- Number: Halide content
- 0: less than 0.05% in weight (“halide-free”)
- 1: halide content depends on activity:
- less than 0.5% for low activity
- 0.5% to 2.0% for moderate activity
- greater than 2.0% for high activity
Any combination is possible, e.g. ROL0, REM1 or ORH0.
J-STD-004 characterizes the flux by reliability of residue from a surface insulation resistance (SIR) and electromigration standpoint. It includes tests for electromigration and surface insulation resistance (which must be greater than 100 MΩ after 168 hours at elevated temperature and humidity with a DC bias applied).
MIL-F-14256 and QQ-S-571[edit]
The old MIL-F-14256 and QQ-S-571 standards defined fluxes as:
| R | (Rosin) |
| RMA | (Rosin mildly activated) |
| RA | (Rosin activated) |
| WS | (Water-soluble) |
Any of these categories may be no-clean, or not, depending on the chemistry selected and the standard that the manufacturer requires.
See also[edit]
References[edit]
- ^"The use of ... borax ... traced back to the ancient Egyptians, who used it as a metallurgical flux". Britannica.com. Archived from the original on 2012-01-14. Retrieved 2011-08-19.
- ^Bhardwaj, Hari C. (1979). Aspects of Ancient Indian Technology (use of lime as a flux). Motilal Banarsidass. ISBN . Archived from the original on 2017-11-03. Retrieved 2011-08-19.
- ^"Metallurgy in southern South America, Smelting, p. 1659-60"(PDF). Archived from the original(PDF) on October 10, 2010. Retrieved 2011-08-19.
- ^"What Is Solder Flux And How Do You Use It?". www.pcbgogo.com. Retrieved 2021-07-09.
- ^"What is Solder and its Types". bestsolderingirons. 2019-12-18. Retrieved 2021-08-05.
- ^"How to Use Flux When Soldering Electronics For Beginners". Solderingironguide. 2019-12-18. Retrieved 2021-07-09.
- ^"Why use flux when soldering?". Engineering and Component Solution Forum - TechForum │ Digi-Key. 2019-07-03. Retrieved 2021-07-09.
- ^"Society of American Silversmiths". Silversmithing.com. Archived from the original on 2010-12-01. Retrieved 2010-03-02.
- ^"FAQ on fluorides in flux". Fluoridefreeflux.com. Archived from the original on 2011-07-20. Retrieved 2011-08-19.
- ^Shangguan, Dongkai (2005). Lead-free solder interconnect ... - Google Books. ISBN . Archived from the original on 2013-06-20. Retrieved 2011-08-19.
- ^""colophony disease", Archaic Medical Terms List, Occupational, on Antiquus Morbus website". Antiquusmorbus.com. 2011-07-29. Archived from the original on 2011-09-03. Retrieved 2011-08-19.
- ^Controlling health risks from rosin (colophony) based solder fluxes, IND(G)249L, United Kingdom Health and Safety Executive, 1997 (online PDF)Archived 2011-01-12 at the Wayback Machine
- ^Humpston, Giles; Jacobson, David M. (2004). Principles of soldering - Google Books. ISBN . Archived from the original on 2013-06-20. Retrieved 2011-08-19.
- ^"Flux-Coated Solder Preforms". Indium.com. 2011-08-15. Archived from the original on 2011-07-19. Retrieved 2011-08-19.
- ^Humpston, Giles; Jacobson, David M. (2004). Principles of soldering - Google Books. ISBN . Archived from the original on 2013-06-20. Retrieved 2011-08-19.
- ^Electronic Materials Handbook: Packaging - Google Books. November 1989. ISBN . Archived from the original on 2013-06-20. Retrieved 2011-08-19.
- ^ abLau, John H. (31 May 1991). Solder joint reliability: theory and ... - Google Books. ISBN . Archived from the original on 2013-06-20. Retrieved 2011-08-19.
- ^Popular Mechanics - Google Books. Hearst Magazines. May 1926. Archived from the original on 2013-06-20. Retrieved 2011-08-19.
- ^Judd, Mike; Brindley, Keith (1999-03-31). Soldering in electronics assembly - Google Books. ISBN . Archived from the original on 2013-06-20. Retrieved 2011-08-19.
- ^US Patent 3988175, Soldering flux and methodArchived 2016-04-10 at the Wayback Machine. Baker, James C.; Bauer, Robert E.
- ^"An Investigation of the Chemistry of Citric Acid in Military Soldering Applications"(PDF). 1995-06-19. Archived(PDF) from the original on March 15, 2020.
- ^"Resources Recovered Calculator". Weld Engineering Co. Archived from the original on 15 May 2015. Retrieved 5 March 2015.
- ^"Din en 29454-1:1994-02". Archived from the original on 2016-02-06. Retrieved 2016-02-06.
- ^"Archived copy"(PDF). Archived(PDF) from the original on 2013-11-06. Retrieved 2013-10-14.: CS1 maint: archived copy as title (link)
External links[edit]
Meaning, and uses of Aquamarine
The healing properties of aquamarine are ideally suited for emotional, spiritual, and physical healing. The soothing and calming effects of the stone, indicated in its meaning, help uncover the underlying anger and fears that are the cause of all emotional trauma and help deal with them in a truthful and meaningful way. If there's an old emotional trauma preventing you from moving forward in life, aquamarine’s cleansing properties will make things easier to let go of.
The damaging of one’s ego can cause that person to find themselves inadequate and undeserving of true happiness, often leading them to abusive relationships and toxic friendships. Aquamarine’s powers let us see the true nature of our situation more clearly and act accordingly. It is possible to begin your emotional healing even if you’ve allowed yourself to be a martyr for too long; once your inner truths are revealed to you true aquamarines meaning, your manipulative relationships will be easier to deal with once and for all.
The first step in any emotional growth is recognizing the negative patterns of behavior that led you to the situation you might be in. The healing properties of the blue gemstone also facilitate communication with others. It is easier to work out any disagreements, with less anger and fear, as indicated earlier, when we talked about the stone’s meaning. The benefits of aquamarine allow you to communicate with compassion, more rationally, and intelligently approaching every conflict and disagreement.
Aquamarine can benefit children as well, especially those who have endured trauma in the past and put up emotional barriers and exhibit aggressive behavior as a result of said trauma. Parents often cause damage with unrealistic expectations and a judgmental approach. An aquamarine can be of massive help in healing that damage in children and adults. Commonly this sort of emotional trauma is the root of anxious feelings and panic attacks caused by guilt and a sense of inadequacy. All of these issues can be dealt with successfully using aquamarine.
For the best results in handling emotional issues, carry an aquamarine worry stone and hold it during times of stress. Aquamarine jewelry, such as earrings and necklaces are also helpful since it is recommended to keep the gems close to your head and neck.
The properties of aquamarine related to physical healing are thought to be closely connected with breathing. Sometimes referred to as the “breath stone,” aquamarine is known to alleviate sinus, lung, and respiratory problems. It is also believed to help with bronchitis, colds, hay fever, and various allergies. In terms of other issues, like conditions and diseases of the skin, aquamarine seems to be beneficial for anyone suffering from skin inflammations.
Things like rosacea, psoriasis, hives, and eczema, can be calmed with the soothing benefits of aquamarine stone, which is in keeping with the aquamarine meaning. It can severely reduce or even prevent herpes outbreaks and can help with shingles as well when used in tandem with regular therapy. Laryngitis and sore throats can be soothed by aquamarine, due to its “cooling stone” properties.
Commonly teeth and gums can be alleviated, too, with proper use. It is thought to encourage optimum growth and hormone production from the pituitary and thyroid glands. To achieve the best results when dealing with physical issues, wear aquamarine jewelry near the afflicted area of your body or place gently cooled gemstones directly on the area in question.
If you’re looking to reduce tiredness of the eyes and eye irritation, put aquamarines on your eyelids for twenty to thirty minutes each night before bed. To relieve nervous spasms and heart palpitations, place an aquamarine gemstone just below the middle of your breastbone, on the solar plexus.
Aquamarine’s ability to make people feel stronger and more empowered enhances its spiritual healing properties. However, even though it helps us feel more confident, it also allows us to realize that there are many sources of power other than sheer force. According to aquamarine, meaning, compassionate communication with oneself, trough, honest, and transparent thoughts allows us to go through a journey of self-improvement.
Women tend to find the strength and courage to express their true feelings and ideas, as well as finding it more comfortable to wield their powerful intuition. Men, on the other hand, tend to find it easier to cut through their emotional numbness barrier, which allows for more precise communication through unhindered emotional expression. Taking away the walls and gates of communication makes spiritual healing exponentially easier.
The reflective capabilities of aquamarine enable hidden truths to be revealed, thus leading to self-awareness and empowerment. Aquamarine’s healing properties help us improve our communication with ourselves and each other, but most fundamentally, with the Divine. As supported by aquamarine, meaning, messages, and articulations to the Divine are more lucid and more potent.
Often considered a gateway crystal to spiritual access, aquamarine can help you achieve a closer connection with the outer manifestations of your spirituality and with your inner self. For religious purposes, use aquamarine mala, worry beads or prayer beads, wear pendant earrings or aquamarine necklace, or hold an aquamarine worry stone as you initiate communication with the Divine.
The meaning of aquamarine that relates to spirituality is to go with what life gives you at any given moment, rather than lying in wait for the perfect time or opportunity. Also, dreaming about aquamarine is often interpreted as a sign of a new friendship waiting to happen.
Aquamarine stones can help make the connection one has with their guardian angels stronger. Anyone born between March 21 and March 25 can boost their connection with Vehujah by wearing light aquamarine, whereas those born between July 28 and August 1 can strengthen their relationship to Haaiah through the same practice. In Feng Shui, aquamarine channels water energy. This type of energy is focused on regeneration and rebirth, and it encourages qualities like stillness, quiet strength, and purification.
Put aquamarine near the northern end of your bedroom, study, or whatever area you use the most for prayer, repose, and calm reflection in your home, and you can expect a world of benefits in keeping with aquamarine meaning.
Activation of CO and CO2 on homonuclear boron bonds of fullerene-like BN cages: first principles study
Abstract
Using density functional theory we investigate the electronic and atomic structure of fullerene-like boron nitride cage structures. The pentagonal ring leads to the formation of homonuclear bonds. The homonuclear bonds are also found in other BN structures having pentagon line defect. The calculated thermodynamics and vibrational spectra indicated that, among various stable configurations of BN-60 cages, the higher number of homonuclear N-N bonds and lower B:N ratio can result in the more stable structure. The homonuclear bonds bestow the system with salient catalytic properties that can be tuned by modifying the B atom bonding environment. We show that homonuclear B-B (B2) bonds can anchor both oxygen and CO molecules making the cage to be potential candidates as catalyst for CO oxidation via Langmuir–Hinshelwood (LH) mechanism. Moreover, the B-B-B (B3) bonds are reactive enough to capture, activate and hydrogenate CO2 molecules to formic acid. The observed trend in reactivity, viz B3 > B2 > B1 is explained in terms of the position of the boron defect state relative to the Fermi level.
Introduction
The prospect of utilizing non-metal materials for the adsorption and catalytic conversion of toxic environmental gases, as an alternative for the present-day precious metal catalyst is gaining interest, owing to its lower price as well as a better durablility1,2,3,4,5,6. Among metal-free adsorbents, carbon based nanostructures, such as C60, carbon nanotube (CNT) and graphene have received much attention7,8,9. Similar interest is directed to, BN analogue: it was discussed that with modified electronic structures it can also lead to promising materials for gas capturing and catalytic convertors10,11,12,13. The BN based monolayer and nanotube structures have been quite widely studied experimentally as well as theoretically14,15. It is noteworthy that, a recent experimental study has demonstrated the possibility of systematically converting a graphene sheet to a hexagonal BN sheet via a chemical route16. Combining the chemical route with the lithography technique it is possible to produce uniform boron nitride structures without disrupting the structural integrity. Also the carbon based template can be used to synthesize the BN structures17. Inspired by these experiments, in the present work, we explore properties of fullerene-like BN cages, hereafter named as BN-60, which may be obtained as a result of atom by atom substitution of C60 or by direct synthesis. The important point is that the network of pentagonal rings in BN-60 will lead to homonuclear bonds18. The BN cages, free of the homonuclear bonds, are made up of square and hexagon rings as discussed in previous literature19,20,21. However, pentagon–octagon–pentagon line defects are found in the BN sheets, nanoribbons and single-walled BN nanotubes and are consequence of the existence of homonuclear bonds22. Under boron rich environment the large possibility of formation of frustrated B-B homonuclear bond has been reported23. Also the pentagons with homonuclear bond form at the tip of the h-BN nanotube24. In the present work, we found that the homonuclear bonds have decent reactivity, which is distinctly different from the conventional BN structures. We are particularly interested in the catalytic performance of homonuclear bonds for CO oxidation and CO2 conversion.
The oxidation of CO is an important prerequisite for mitigating toxic CO gas. On a catalyst surface, CO oxidation follows Langmuir–Hinshelwood (LH) mechanism and the Eley–Rideal (ER) mechanism. LH mechanism involves the coadsorption of reactants onto the catalytic surface, followed by a surface reaction to form the products. ER mechanism, on the other hand, involves the direct reaction of a gaseous reactant with a chemisorbed one. Nitrogen-doped carbon nanotubes possess the ability to effectively catalyze the CO oxidation with activation energies ranging from 0.477 to 0.619 eV. A less negative charge on the dopant N atom is correlated with a higher activity for CO oxidation25. Iron embedded graphene also proved to be a potential material for CO oxidation with activation energy of 0.58 eV26. Graphene doped with Cu results in electronic resonance among the electronic states of the reactants and the Cu atom, leading to higher reactivity for oxidizing CO. The process proceeds first via an LH mechanism with barriers of 0.25 eV and 0.54 eV followed by ER reaction without energy barrier27. Zhao et al. have investigated theoretically the possibility of CO oxidation on a Si embedded graphene surface and attributed to the charge transfer from the embedded Si atom to the 2π* orbital of O2. The process proceeds first via LH mechanism with a barrier of 0.48 eV followed by ER mechanism28. Fe encapsulated boron nitride cage has good CO to CO2 conversion capabilities with an activation energy of 0.5 eV29. The choice of dopants significantly alters the CO oxidation mechanism and hence the activity of boron nitride monolayer. This was demonstrated in our previous work employing carbon and oxygen as dopants, wherein the O dopant enabled chemisorption of CO, while C doped h-BN monolayer has lesser tendency to adsorb CO30. O doping results in a larger bond length of a neighboring B atom, it’s out of plane displacement and less positive charge, synergistically contributing to stronger CO adsorption.
Metal-organic frameworks, Carbon and BN nanostructures, such as CNT and BN nanotube (BNNT)31,32,33, have also been tested for CO2 capture and storage. As the weak binding of CO2 on such inert surfaces is a demerit, various methods to activate the surface have been tested. Suchitra et al. reported that Boron doped C60 (BC59) fullerene does not adsorb CO2 molecule effectively but 1e− charged BC59 can strongly adsorb CO2 with binding energy of −0.66 eV31. Huang et al. proposed about the remarkable CO2 capturing ability of armchair graphene nanoribbons with dangling bond defect, the adsorption energy is about −0.31 eV34. Sun et al. provided a route to increase the activity of a pure BN sheet to adsorb CO2 by applying the electric field. By applying 1.36 eV of electric field the adsorption energy can be increased to −0.84 eV35. Gao et al. demonstrated that single Ca atom anchored on C60 can adsorb CO2 with higher binding energy compared to pristine C6036. Shao et al. proposed the increase of chemical activity of BNNT by the substitutional doping of Al atom in-place of B site. The CO2 binding energy varies with tube diameter and is in the range of about −0.03 to −5.08 eV37.
In the present work, the BN analogues of C60, which possess the frustrated homonuclear bonds because of pentagons, are investigated in the perspective of aforementioned CO oxidation and CO2 conversion catalyst. The stability of the cages has been analyzed and discussed in detail. It has been discoursed how the pentagonal rings in the structure will generate the homonuclear B-B, B-B-B, N-N and N-N-N bonds. Throughout this work, we designate B1 notation for a single B atom surrounded entirely by nitrogen, B2 for two B atoms bonded together, making a B-B bond and B3 for two adjacent B-B bonds merged to form a B-B-B bond for simplicity. Similarly for N sites we consider the similar notation. The binding affinity of the different stable BN-60 cages considering B1, B2 and B3 sites to capture CO/CO2/O2 molecules has been estimated. The role of the sites (B1, B2 and B3) on the CO oxidation and CO2 conversion are analyzed in detail using first principles approach.
Results and Discussions
Construction of BN-60 cages
Here we first outline the scheme adopted for the replacement of carbon atoms in C60 cage, containing 12 pentagonal and 20 hexagonal rings with B and N atoms to construct the BN-60 cages. In general, there are many ways to construct the BN-60 cages containing different distribution of B and N atoms, B/N ratio and homonuclear B and N bonds. In this work we made three different classes of BN-60 cages, considering 1. Boron rich, 2. Nitrogen rich and 3. stoichiometric B:N environment. To make the BN-60 cages with these environments, our approach is to make homonuclear B and N bonds first, taking into account pentagonal rings and then following some rules. Initially, replacement of carbon atoms is done on pentagonal rings such that each pentagonal ring has one homonuclear bond of either type (B2 or N2). In fact, it is evident from recent experiments that the presence of homonuclear bonds on pentagonal rings of boron nitride structures are inevitable20. Another important consideration is that we restrict the homonuclear bonds to a maximum of three B (B3) and three N (N3) atoms. So to make B rich cages, all pentagonal rings are filled by B2 bonds and each B2 is surrounded by three or four homonuclear B2 bonds with one carbon atom separating them. This constraint prevents the formation of homonuclear N2, B3 or N3 configurations. The remaining carbon atoms in both hexagonal and pentagonal rings are replaced by B and N atoms alternatively, to avoid the N2 bonds. To incorporate B3 bonds in the boron rich condition, two or three B2 configurations should be surrounded by two or three B2 bonds.
The N rich BN-60 cages are constructed in a similar fashion incorporating N2 bonds instead of B2 bonds. In order to make stoichiometric B:N type BN-60 cages, the 12 pentagonal rings should be equally shared by both B2 and N2 bonds (6 B2 and 6 N2). Arrange these B2 and N2 bonds by alternative or continuous way first so that the number of B2 and N2 bonds is same and now depending on the surrounding homonuclear bonds, rest of the carbon atoms in both hexagonal and pentagonal rings are replaced by both B and N atoms. The steps for constructing stoichiometric 1-B30N30 type cage with 5 B2, 2 B3 & 5 N2, 2 B3 bonds is demonstrated in the Fig. S1 of the supplementary information (SI). This approach would allow us to consider the effects of homonuclear bonds that are always likely to occur on pentagonal rings of BN nanostructures.
Stability of BN-60 cages
In order to analyze the stability of different BN-60 structures, the formation enthalpy (F.E) per atom has been estimated using the following equation,
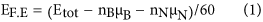
where, Etot is the total energy of the different type of homonuclear bonded BN-60 structure. nB is the number of boron atoms replaced the carbon atoms; μB is the chemical potential of the boron atom (reference structure: α-rhombohedral phase of bulk boron); nN is the number of nitrogen atoms replaced the carbon atoms; μN is the chemical potential of the nitrogen atom (reference structure: N2 gas molecule). Higher negative values of formation enthalpy for BN-60 cages, as obtained from equation (1), indicate better stability of the system.
In Table 1, the formation enthalpy values for different type of homonuclear bonded BN-60 cages are summarized. In every case the EF.E is negative which clearly indicates the stability of the systems. The formation enthalpy value is plotted against the configuration of the system and shown in Fig. 1b and it helps to obtain a clear idea about the relationship between the formation enthalpy and type of BN-60 cage. Among all, N rich systems show a higher negative value than others, hinting that N- rich cages are more favorable to be synthesized in normal pressure and temperature. It is also evident that higher number of B3 bonds compared to the B2 bonds leads to lower stability of the systems, which can be easily understood comparing the formation enthalpy of three different types of B30N30 cages (see Table 1). Based on the formation enthalpy and considering homonuclear B bonds, four BN-60 cages (one B rich, two N rich and one stoichiometric B:N cases) are selected for further calculation and are shaded in grey in Table 1. Geometry optimization for the different type of BN-60 cages was performed and the optimized structure is shown in Fig. 1a and Fig. S2. The B rich or more B bonds in the system leads structural distortion and it is visible in Fig. S2 and Fig. 1a, so it may act as a good medium for gas adsorption. We estimated the density of phonon states (DOPS) for all the BN-60 cages. No negative frequency states because of structural instability have been found. Here only DOPS of four structures are shown in Fig. S3 (B25N35, 1-B27N33, 1-B30N30, B34N26). This indicates that all the structures are highly stable. The DOPS for all the other structures are not shown here.
Full size table
Stability of BN-60 cages.
(a) Relaxed structure of BN-60 cages with different B:N ratio and homonuclear bonds. (b) Formation enthalpy per atom of various BN-60 cages. Blue and pink sphere denoted the N and B atoms.
Full size image
Density of states (DOS)
Next, we gain an understanding on the electronic properties of BN-60 cages. The densities of states (DOS) as a function of energy (eV) for four specific systems (B25N35, 1-B27N33, 1-B30N30, B34N26) are shown in Fig. 2. These results indicate that both B and N atoms in every structure contribute to the valence band maximum and conduction band minima. The contribution vary based on the B:N ratio in the BN-60 cages. For example in case of B25N35, N atoms mainly contribute valence bands whereas the B atoms contributed to the conduction band mostly. The B atoms‘ contribution to the valence bands increases in case of B34N26 cages. Also the B:N ratio and homonuclear B2, B3, N2 and N3 configuration play a major role for generating defect sates. In particular, the DOS for a 1-B30N30 has defect states very near to the Fermi level because of the higher number of B3 and N3 configuration in the system. More understanding about the role of B1 B2 and B3 configuration is discussed in the next section.
Electronic properties of BN-60 cages representing contribution of B and N atoms.
Partial density of states (PDOS) of (a) B25N35 (b) 1-B27N33 (c) 1-B30N30 and (d) B34N26 cages. Black solid line, green dash line and red dotted lines represent the PDOS for total (B and N), only N and only B atoms. Fermi level is consider at 0 eV.
Full size image
Adsorption of molecules
In order to test the catalytic capabilities of the BN-60 cages, we first study the adsorption of molecules, O2, CO and CO2. The high negative charge of nitrogen prevents the adsorption of any of the reactants on N1, N2 and N3 sites30,38. Table 2 shows the calculated adsorption energies of the molecules on the three different boron sites. It can be seen that the reactivity of boron atom strongly depends on its environment. As expected, B1 sites show no evidence of adsorbing any reactant molecules, rendering them unsuitable for catalytic applications. On B1 sites, the O2 adsorption energy (Ead(O2)) varies from 0.001/−0.057 eV (considering PBE/PBE-D functional) in 1-B27N33 to −0.094/−0.198 eV in B25N35, CO adsorption energy (Ead(CO)) varies from 0.003 eV/−0.070 eV in B25N35 to 0.022 eV/−0.337 eV in B34N26 and CO2 adsorption energy (Ead(CO2)) is rather very small in all the cases. Interestingly, B2 atoms are more reactive, with Ead(O2) ranging from −2.492/−2.622 in 1-B30N30 to −2.854/−2.988 in B25N35, Ead(CO) varying from 0.028 eV/−0.344 eV in B34N26 to −0.539/−0.652 eV in 1-B27N33 and Ead(CO2) varying from 0.052/−0.079 in 1-B27N33 to −0.195/−0.375 eV in B25N35. The optimized geometries of B27N33 after the adsorption of molecules are shown in Fig. S4. The high affinity toward O2 and CO combined with weak CO2 adsorption suggest that B2 sites of BN-60 cages can be efficient in catalyzing CO oxidation, which is discussed in detail later. The B3 sites strongly adsorb the O2, CO and CO2 molecules, with (Ead(O2)) ranging from −2.903/−3.043 eV in 1-B27N33 to −3.470/−3.599 eV in 1-B30N30, Ead(CO) ranging from −1.185/−1.294 in 1-B27N33 to −1.678/−1.784 eV in B34N26 and Ead(CO2) varying from −0.196/−0.382 eV in 1-B27N33 to −1.214/−1.361 eV in B34N26. The ability of B3 sites to anchor CO2 molecules is of particular interest as these systems can be employed as metal-free CO2 trapping agents to solve many environmental problems. On adsorption, the CO2 molecule is strongly activated as is evident from the bent geometry of the molecule (Fig. S4).
Full size table
Bader charge analysis also confirms that the B3 sites are the highest reactive sites. The net charges are given in units of e, with a positive charge indicating a deficit of charge and a negative charge indicating a surplus of charge. The Bader charge analysis is performed, taking 1-B27N33 as a representative case. Before adsorption, B1 has a net charge of +2.129. After the adsorption of CO, O2 and CO2 on 1-B27N33, the net charge on B1 is +2.146, +2.145 and +2.135 respectively (given in Table 3), indicating that there is very little charge transfer to the incoming molecules. The net charges on the two atoms constituting B2 changes from +1.344, +1.441 before adsorption to +1.423, +1.577 on CO adsorption, +2.19, +2.16 after O2 adsorption and +1.319, +1.469 on CO2 adsorption and are given in Table 3. These values indicate that the B2 sites are able to donate electrons to the O2 and CO molecules whose net charges post adsorption are −1.545 and −0.268 respectively while the CO2 molecule is unaffected and retains its neutral charge. The three atoms constituting the B3 sites have charges of +1.25, +0.880 and +1.26 before the adsorption of any molecules which changes to +1.519, +0.949 and +1.411 on CO adsorption; +1.414, +1.378 and +2.16 on O2 adsorption and +1.405, +1.376 and +1.976 on CO2 adsorption. The charges on O2, CO and CO2 in this case are −1.54, −0.387 and −1.418 respectively. A point to note is that the charge transfer to the adsorbates is the largest when the boron sites before adsorption have less positive charge and hence more electron density. The central atom of the B3 site hence has the strongest ability to adsorb the incoming molecules.
Full size table
In order to understand the origin of the observed trend in reactivity, viz., B3>B2>B1, we plot the partial density of states (PDOS) of B1, B2 and B3, taking 1-B27N33 as an example as shown in Fig. 3. Inspection of the PDOS reveals that the occupied defect states of B3 sites lie near to the Fermi level (here normalized to lie at 0 eV), indicating their ability to easily transfer electrons to the reactant molecules. Also, the largest contribution to the B3 states comes from the central atom of B3.The defect valence band states of B2 sites are a little further away relative to the Fermi level, while the B1 states are far away from the Fermi level. Thus the position of the defect states because of the homonuclear configuration relative to the Fermi level governs the reactivity.
The partial density of states of the three different active boron sites of 1-B27N33.
Red dotted line indicates B1, green dashed line indicates B2 and black solid line indicates B3 PDOS. Fermi level is considered to lie at 0 eV.
Full size image
To gain more insight on the reactivity of B2 site and B3 sites, we calculated the charge density difference (CDD) upon the CO and O2 adsorption on 1-B27N33, as depicted in Fig. 4. The isovalue is set at 0.005 e/Bohr3. The yellow and blue lobes represent the charge accumulation and the charge depletion, respectively. The CDD plots well explain that the B3 sites are more active than the B2 sites in interacting with an incoming molecule. All the plots demonstrate that O2, CO and the surface undergo considerable charge redistribution on adsorption: the molecules acquire electrons from B27N33. The depletion of charge in the O-O and C-O bond regions of O2 and CO imply that the molecules are strongly bound to the surface, resulting in the elongation of the intramolecular bond. The charge depletion from both the atoms of the B2 sites upon the O2 adsorption can be clearly observed in the plot in Fig. 4(a). In addition, in the case of B3 sites (see Fig. 4(b)), the bond connecting the third B atom, which is indicated by an arrow and is not directly bonded with the incoming molecule, suffers from some amount of charge depletion, indicating that this site also plays a role in donating electrons to the incoming molecule. The observed trend of the higher reactivity of B3 site can be attributed to the overall charge donation feature of B3 atoms. The same observation is also found during CO adsorption. Even though CO molecule is attached to a single boron atom of either B2 or B3 (see Fig. 4(c,d)), the other boron atoms (indicated by arrows) constituting the homonuclear bonds also take part in electron donation to the incoming molecule, promoting the overall binding ability and hence B3 sites adsorb the strongest. This feature is also evident from Bader charge analysis, tabulated in Table 3, wherein all the boron atoms constituting the homonuclear bonds lose electronic charge on interacting with either CO or O2.
Charge density difference for O2 and CO adsorption.
O2 anchored on (a) B2 site of B27N33, (b) B3 site of B27N33 and CO anchored on (c) B2 site of B27N33, (d) B3 site of B27N33. Blue and yellow lobes correspond to a depletion and accumulation of electronic charge, respectively. The isosurface value of 0.005 e/Bohr3 is considered for all the cases. The arrows indicate the boron atom/s in the active site not bonded with the adsorbate. Pink, blue, red and gray balls indicate B, N, O and C atoms, respectively.
Full size image
Reactivity of homonuclear bond
The PDOS of the boron sites (B1, B2, B3) after the adsorption of molecules is shown in Fig. 5. It can be seen from Fig. 5a,d and g that there is no charge transfer from the B1 sites to any of the incoming adsorbate molecules. The molecular orbitals of O2, CO and CO2 retain their isolated characteristics, depicted in Fig. 5. Figure 5b,c show the PDOS of B2 and B3 sites and the O2 molecule upon adsorption. The spin-up π* (O2) states lie just above the Fermi level and can act as acceptor levels39. In fact, upon adsorption on B2 and B3 sites, these states become occupied, shift downward and hybridize with the p states of B2 and B3 atoms, explaining the higher interaction. Figure 5e,f demonstrate the DOS of B2, B3 and CO molecule after adsorption. The antibonding (π*) states of CO lie at around 2 eV above the Fermi level. As the valence states of B2 and B3 are closer to the LUMO of CO, the interaction of the CO molecule with B2 and B3 sites results in charge transfer to π* orbital, resulting in stronger adsorption.
PDOS to explain the interaction behavior of the gas molecules with active boron sites.
Partial density of states of (a–c) O2 on B1, B2 and B3 sites respectively; (d–f) CO on B1, B2 and B3 sites respectively; (g–i) CO2 on B1, B2 and B3 sites respectively of 1-B27N33 after adsorption. Fermi level is consider at 0 eV.
Full size image
The CO2 molecule does not interact with the B2 sites,as can be seen from Fig. 5h. The chemisorption of CO2 onto the B3 sites takes place in two steps: the bending of the CO2 molecule followed by its adsorption40,41. In order to understand this mechanism, we first performed a density of states analysis of an isolated bent CO2 molecule, keeping the bond lengths and bond angles same as the adsorbed configuration (see Fig. S5). The density of states clearly illustrates the splitting of the HOMO (1π) and LUMO (2π*) orbitals of the CO2 molecule into two states42. The split LUMO orbitals are named 2a and 2b, of which 2b can now readily accept electrons as it lies closer to the Fermi level. This can be understood by inspecting the DOS of B3 and CO2 after their interaction (Fig. 5i), wherein the 2b states become occupied, resulting in the weakening of C-O bond of CO2 and hence stronger adsorption. Based on the estimated results and analysis, we can conclude that for complete CO oxidation B2 sites are more suitable and for CO2 capturing and conversion B3 sites are superior.
CO oxidation and free energy profile
Now we investigate the mechanism by which CO oxidation occurs on B2 site of 1-B27N33 and B25N35. This site is able to anchor both CO and O2 implying that the CO oxidation may follow the LH mechanism. The free energy profile is constructed by taking ΔG = ΔE – TΔS + ΔZPE, where ΔE is the total energy change obtained from DFT calculations, ΔS denotes the entropy change and ΔZPE is the change in the zero point energies. TS of free molecules are obtained from ref. 43, while TS of the adsorbates and ZPE of the free molecules and adsorbates are estimated from the DFT calculations considering vibrational frequencies of the molecules in the harmonic approximation, freezing the BN cage44. The ZPE correction is calculated as ZPE = ½∑iћωi, where ћ is the reduced Planck’s constant and ωi is the frequency of the ith vibrational mode of the adsorbate molecule. The entropic term of the free energy is calculated from:
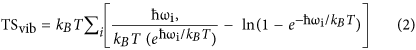
where 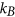 denotes the Boltzmann constant.
denotes the Boltzmann constant.
The images demonstrating the reaction steps of CO oxidation via LH mechanism is shown in Fig. S6. The initial step of LH mechanism is taken to be the one in which 2CO molecules and an O2 molecule are far from the surface and do not interact. The co-adsorption of CO and O2 on the B2 site is taken to be the next step and the optimized structure is shown in the inset of Fig. 6. The O2 molecule which was initially in the triplet state loses its magnetic moment upon adsorption. Detailed information on the changes in the spin state of molecular oxygen upon adsorption is explained in the supporting information and tabulated in Table S1. The desorption of first CO2 from the surface requires an activation energy (Ea) of around 1.14 eV in 1-B27N33 and 1.35 eV in B25N35. This step is hence the rate limiting step with the highest activation barrier. The overall reaction is exothermic (ΔG = −5.18 eV in 1-B27N33 and −5.16 eV in B25N35) and the remaining O atom migrates toward the epoxy site. The O atom then readily reacts with another incoming CO molecule to generate the next CO2 molecule. This reaction requires that a thermodynamic barrier of 1.06 eV in 1-B27N33 and 1.08 eV in B25N35 be surmounted. The calculated values of Ea and adsorption energies of reactants are used to estimate the Sabatier activities of CO oxidation over B27N33 and B25N35. The SA can be used as a measure of the ability of the catalyst to catalyze the process of CO oxidation. The first reaction step (R1 of SI) is taken as the one in which CO is adsorbed and in the next step the O2 molecules adsorb (R2 of SI) on neighboring active sites. This results in the formation of a (O2···CO)* intermediate. The activation barrier for desorption of first CO2 from this intermediate plays a decisive role in the overall activity. Also we found that the very high binding strength of molecules on the surface influences the activity. The Sabatier activities of 1-B27N33 and B25N35 are found to be −1.3 and −1.8 respectively. We have also calculated the SA of B30N30 and found it to be −0.61. The reaction rate is influenced by both the temperature and activation energies for CO oxidation. For instance, in the LH mechanism, after CO adsorption, O2 adsorbs in a neighboring site, forming a (O2···CO)* intermediate. We have considered the removal of first CO2 from this intermediate to be the rate determining step (R3 in the SI), because of the high activation barrier. The Arrhenius equation is:
Free energy pathways of CO oxidation via LH mechanism on (a) 1-B27N33 and (b) B25N35. The lower left insets show the co-adsorption of O2 and CO on B2 site, which is the initial state, while the upper right insets show the final state for the formation of first CO2. The initial state and final states are denoted as I.S. and F.S. Ea denoted the activation barrier. The *sign indicates the catalytic surface and the adsorbed state of molecules and atoms are denoted with a *sign. Blue, pink, grey and red spheres denote the N, B, C and O atoms. The dotted lines are to guide the eye.
Full size image
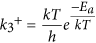
where 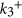 is the rate constant. Thus, the higher the temperature, the easier it is for the reactants to surmount the activation barrier45. In this work, the calculations of the rate constants, rate and the Sabatier activity are performed at a temperature of 273 K. At higher temperatures, the activation processes are expected to be faster. The detailed reaction steps and calculation procedure are outlined in the supplementary information. We have also compared the calculated Sabatier activities of the BN nanocages with a few other conventional catalysts available in the literature and found that B30N30 cage performs good, showing the excellent activity (see Table S2 of supporting information).
is the rate constant. Thus, the higher the temperature, the easier it is for the reactants to surmount the activation barrier45. In this work, the calculations of the rate constants, rate and the Sabatier activity are performed at a temperature of 273 K. At higher temperatures, the activation processes are expected to be faster. The detailed reaction steps and calculation procedure are outlined in the supplementary information. We have also compared the calculated Sabatier activities of the BN nanocages with a few other conventional catalysts available in the literature and found that B30N30 cage performs good, showing the excellent activity (see Table S2 of supporting information).
CO oxidation on boron nitride nanotube with defects
The presence of similar kind of homonuclear B-B bonds have been observed in defective BN nanosheets and nanotubes. In particular the Stone-Wales defect (SW) in the BN based system has been studied theoretically46. Also, spectroscopic studies suggest the existence of such defects is more feasible in the boron nitride nanotubes47. A recent atomic resolution imaging study has also confirmed the presence of Stone-Wales like defects in boron nitride sheets48. To investigate the activation processes on B-B sites, we take an example of a SW defect on a BNNT (henceforth named as SW-BNNT). To model this system, we have chosen a (7, 0) supercell consisting of 42 BN formula units. The SW defect is formed by rotating a BN bond by 900. The O2 molecule adsorbs at a B-B bond in the side-on fashion with adsorption energy of −2.97 eV, while CO adsorbs at the boron site of a B-B bond with adsorption energy of −0.08 eV, at a distance of 1.62 Ǻ from the surface. The adsorption energies of O2 and CO on SW-BNNT are stronger than those on a pristine BNNT. This is in agreement with previous results49. In order to compare the activity with the nanocages, we consider only the LH mechanism, wherein the CO molecule is adsorbed first followed by the adsorption of O2, to form a CO···O2 intermediate (see Fig. 7 for the entire energy profile). The removal of first CO2 requires an activation barrier of 1.23 eV, leaving behind an oxygen atom in the epoxy position. The high value of Ea can be justified based on the weak adsorption of CO molecule unto the surface, implying that O2 bond breaking is difficult. The reaction is exothermic by −5.23 eV. The reactive O atom then interacts with another CO molecule to form the second CO2 molecule. We note in passing that CO oxidation via ER mechanism is also probable and may take place requiring a smaller activation barrier as the O2 molecule adsorbs quite strongly in the side on fashion.
Free energy pathways of CO oxidation on SW-BNNT via LH mechanism.
The initial and final states for the desorption of first CO2 are shown in the lower and upper insets respectively. Ea denotes the activation barrier. The sign conventions and atom colors are similar to that followed in Fig. 6. The dotted lines are to guide the eye.
Full size image
CO2 conversion
As mentioned earlier, the B3 sites are able to capture CO2 effectively in the BN cages. We examine here the possibility of effectively hydrogenating this activated CO2 into formic acid, which is widely used as a chemical fuel. We have taken two examples of BN cages, namely 1-B27N33 and B30N30 to test the photocatalytic CO2 conversion capabilities via a COOH mediated mechanism50. The free energy profile for this process is shown in Fig. 8. We use similar convention to find the free energy change (ΔG) as discussed in previous section. Here it has been assumed that ‘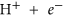 ’ is in equilibrium with
’ is in equilibrium with 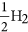 , at pH = 0 and 0 V vs standard hydrogen electrode (SHE)6,51 Initially the CO2 molecule is considered to be far from the surface. The next step involves the adsorption of CO2 onto the B3 sites, which is endothermic in 1-B27N33 by 0.18 eV (Fig. 8a). This is expected as the CO2 molecule does not bind very strongly to 1-B27N33. But in the case of B30N30 (Fig. 8b), the adsorption of CO2 is stronger and hence the first step is exothermic (ΔG = −0.46 eV). The next step, which is the hydrogenation of the activated CO2 at its oxygen atom to form carboxyl (COOH), is uphill by 0.3 eV in 1-B27N33 and 0.73 eV in B30N30. The third step wherein the carbon atom of COOH is attacked by a hydrogen atom to form adsorbed formic acid is mildly endothermic by 0.14 eV in 1-B27N33 and endothermic by 0.10 eV in B30N30. Finally, the adsorbed product, HCOOH desorbs from the surface, with ΔG values being 0.34 eV and 0.60 eV in the case of 1-B27N33 and B30N30 respectively. The low endothermicity of the reaction steps occurring on the B3 sites of 1-B27N33 suggests an exciting possibility of hydrogenating CO2 at near-room temperatures.
, at pH = 0 and 0 V vs standard hydrogen electrode (SHE)6,51 Initially the CO2 molecule is considered to be far from the surface. The next step involves the adsorption of CO2 onto the B3 sites, which is endothermic in 1-B27N33 by 0.18 eV (Fig. 8a). This is expected as the CO2 molecule does not bind very strongly to 1-B27N33. But in the case of B30N30 (Fig. 8b), the adsorption of CO2 is stronger and hence the first step is exothermic (ΔG = −0.46 eV). The next step, which is the hydrogenation of the activated CO2 at its oxygen atom to form carboxyl (COOH), is uphill by 0.3 eV in 1-B27N33 and 0.73 eV in B30N30. The third step wherein the carbon atom of COOH is attacked by a hydrogen atom to form adsorbed formic acid is mildly endothermic by 0.14 eV in 1-B27N33 and endothermic by 0.10 eV in B30N30. Finally, the adsorbed product, HCOOH desorbs from the surface, with ΔG values being 0.34 eV and 0.60 eV in the case of 1-B27N33 and B30N30 respectively. The low endothermicity of the reaction steps occurring on the B3 sites of 1-B27N33 suggests an exciting possibility of hydrogenating CO2 at near-room temperatures.
Free energy pathways of CO2 hydrogenation on (a) 1-B27N33 and (b) B30N30. (c) Optimized structures for each reaction step considering 1-B27N33 cage. The dotted lines are to guide the eye. The sky blue spheres represent hydrogen atom. The other colored spheres represent the same atoms as depicted in Fig. 6. The free hydrogen is not shown in the images for simplicity.
Full size image
Conclusions
We design three different classes of BN-60 cages analogue to C60 cage considering 1. B rich, 2. N rich and 3. stoichiometric B:N environments. The pentagonal rings developed the homonuclear B and N bonds in the BN-60 structures. The formation enthalpy per atom for different configurations has been estimated and found to be in the range of −0.322 to −0.619 eV. N rich BN-60 cages are more favorable compared to other environments and the stability of BN-60 cage with more B2 configuration is higher compared to cages with B3 configuration. The DOPS confirms the good stability of the BN-60 cages. We found that the ability to anchor gas molecules follows B3 > B2 > B1. This trend has been explained considering position of the defect state relative to the Fermi level (B2 > B3). The stronger adsorption of the O2 and CO molecule on B2 and B3 sites is primarily because of charge transfer to π* orbital from the surface states. Only B3 sites can adsorb CO2 molecule, through bending of CO2 molecule, which results in the splitting of LUMO orbitals named 2a and 2b, of which 2b can readily accept electrons. The CO oxidation follows the Langmuir–Hinshelwood (LH) mechanism with Sabatier activity of −1.3 and −1.8 considering 1-B27N33 and B25N35 cage. We found that B3 sites can efficiently convert the CO2 molecule to formic acid. Here we emphasize that the proposed science is a general understanding and will help us to proceed further for an efficient metal free catalyst.
Methods
We performed the calculations using spin polarized density functional theory as implemented in the Vienna ab-initio simulation package (VASP)52. The generalized gradient approximation (GGA) was employed for the exchange and correlation effects at Perdew–Burke–Ernzerhof (PBE)53 and the potentials of the atoms were described by the projected augmented wave (PAW)54 method. For long-range van der Waals attraction the Grimme’s method (DFT-D2) was used with PBE functional (denoted as PBE-D)55. It was found that plane wave cut-off energy of 450 eV was sufficient to get well-converged results. Each BN60 cage was placed in a cubic supercell of size 15 Å, to avoid interactions between periodically repeating images which are at about 9 Å distances. Brillouin zone integration was performed at the Γ point only. All the structures were optimized until the total energy converged to less than 10−5 eV per atom and the maximum force converged to lower than 0.001 eVÅ−1. Density functional perturbation theory (DFPT) has been used to calculate the density of phonon states (DOPS)56. The nudged elastic band (NEB) method was employed to estimate the barrier energy57.
Additional Information
How to cite this article: Sinthika, S. et al. Activation of CO and CO2 on homonuclear boron bonds of fullerene-like BN cages: first principles study. Sci. Rep.5, 17460; doi: 10.1038/srep17460 (2015).
References
Kumar, B. et al. Renewable and metal-free carbon nanofibre catalysts for carbon dioxide reduction. Nature Communications 4, 2819 (2013).
ADSArticle Google Scholar
Chen, Z., Higgins, D. & Chen, Z. Electrocatalytic activity of nitrogen doped carbon nanotubes with different morphologies for oxygen reduction reaction. Electrochimica Acta, 55, 4799–4804 (2010).
CASArticle Google Scholar
Wu, P., Du, P., Zhang, H. & Cai, C. Graphdiyne as a metal-free catalyst for low-temperature CO oxidation. Phys. Chem. Chem. Phys. 16, 5640–5648 (2014).
CASArticle Google Scholar
Shelef, M. Selective Catalytic Reduction of NOx with N-Free Reductants. Chem. Rev. 95, 209–225 (1995).
CASArticle Google Scholar
Qu, L., Liu, Y., Baek, J.-B. & Dai, L. Nitrogen-Doped Graphene as Efficient Metal-Free Electrocatalyst for Oxygen Reduction in Fuel Cells. Acs Nano 4, 1321–1326 (2010).
CASArticle Google Scholar
Shin, D., Sinthika, S., Choi, M., Thapa, R. & Park, N. Ab Initio Study of Thin Oxide–Metal Overlayers as an Inverse Catalytic System for Dioxygen Reduction and Enhanced CO Tolerance. ACS Catalysis 4, 4074–4080 (2014).
CASArticle Google Scholar
Alonso, A. M., Tascón, J. M. D. & Bottani, E. J. Physical Adsorption of Ar and CO2 on C60 Fullerene. J. Phys. Chem. B. 105, 135–139 (2001).
Article Google Scholar
Lu, C., Bai, H., Wu, B., Su, F. & Hwang, J. F. Comparative Study of CO2 Capture by Carbon Nanotubes, Activated Carbons and Zeolites. Energy & Fuels. 22, 3050–3056 (2008).
CASArticle Google Scholar
Wang, L., Ambrosi, A. & Pumera, M. “Metal-Free” Catalytic Oxygen Reduction Reaction on Heteroatom-Doped Graphene is Caused by Trace Metal Impurities. Angew. Chem. Ind. Ed. 52, 13818–13821 (2013).
CASArticle Google Scholar
Mousavi, H., Kurdestany, J. M. & Bagheri, M. Carbon dioxide detection by boron nitride nanotubes. Appl Phys A. 108, 283–289 (2012).
CASADSArticle Google Scholar
Lin, S., Ye, X. & Huang, J. Can metal-free silicon-doped hexagonal boron nitride nanosheets and nanotubes exhibit activity toward CO oxidation? Phys. Chem. Chem. Phys. 17, 888–895 (2015).
CASArticle Google Scholar
Esrafili, M. D. & Nurazar, R. A density functional theory study on the adsorption and decomposition of methanol on B12N12 fullerene-like nanocage. Superlattices Microstruct. 67, 54–60 (2014).
CASADSArticle Google Scholar
Esrafili, M. D. & Nurazar, R. Potential of C-doped boron nitride fullerene as a catalyst for methanol dehydrogenation. comput. Mater. Sci. 92, 172–177 (2014).
CASArticle Google Scholar
Terrones, M. et al. Pure and doped boron nitride nanotubes. Materials today. 10, 30–38 (2007).
CASArticle Google Scholar
Li, R. et al. Non-covalent surface modification of boron nitride nanotubes for enhanced catalysis. Chem. Commun. 50, 225–227 (2014).
CASArticle Google Scholar
Gong, Y. et al. Direct chemical conversion of graphene to boron- and nitrogen- and carbon-containing atomic layers. Nature Communications. 5, 3193, (2014).
ADSArticle Google Scholar
Han, W., Bando, Y., Kurashima, K. & Sato, T. Formation of Boron Nitride (BN) Fullerene-Like Nanoparticles and (BN)xCy Nanotubes Using Carbon Nanotubes as Templates. Jpn. J. Appl. Phys. 38, 755–757 (1999).
ADSArticle Google Scholar
Fowler, P.W., Rogers, K. M., Seifert, G., Terrones, M. & Terrones, H. Pentagonal rings and nitrogen excess in fullerene-based BN cages and nanotube caps. Chemical Physics Letters, 299, 359–367 (1999).
CASADSArticle Google Scholar
Oku, T., Narita, I. & Nishiwaki, A. Synthesis, Atomic Structures and Electronic States of Boron Nitride Nanocage Clusters and Nanotubes. Materials and Manufacturing Processes. 19, 1215–1239 (2004).
CASArticle Google Scholar
Venkataramanan, V. S. et al. Theoretical investigation on the alkali-metal doped BN fullerene as a material for hydrogen storage. Chemical Physics. 377, 54–59 (2010).
CASADSArticle Google Scholar
Wang, Q., Sun, Q., Oku, T. & Kawazoe, Y. First-principles study of La–B36N36 cage. Physica B. 339, 105–109 (2003).
CASADSArticle Google Scholar
Li, X., Wu, X., Zeng, X. C. & Yang, J. Band-Gap Engineering via Tailored Line Defects in Boron-Nitride Nanoribbons, Sheets and Nanotubes. ACS Nano. 6, 4104–4112 (2012).
CASArticle Google Scholar
Yazyev, O. V. & Chen, Y. P. Polycrystalline graphene and other two-dimensional materials. Nature Nanotechnology. 9, 755–767 (2014).
CASADSArticle Google Scholar
Saito, Y. & Maida, M. Square, Pentagon and Heptagon Rings at BN Nanotube Tips. J. Phys. Chem. A. 103, 1291–1293 (1999).
CASArticle Google Scholar
Hu, X., Wu, Y. & Zhang, Z. CO oxidation on metal-free nitrogen-doped carbon nanotubes and the related structure–reactivity relationships. J. Mater. Chem. 22, 15198–15205 (2012).
CASArticle Google Scholar
Li, Y., Zhou, Z., Yu, G., Chen, W. & Chen, Z. CO Catalytic Oxidation on Iron-Embedded Graphene: Computational Quest for Low-Cost Nanocatalysts. J. Phys. Chem. C. 114, 6250–6254 (2010).
CASArticle Google Scholar
Song, E. H., Wen, Z. & Jiang, Q. CO Catalytic Oxidation on Copper-Embedded Graphene. J. Phys. Chem. C. 115, 3678–3683 (2011).
CASArticle Google Scholar
Zhao, J.-X., Chen, Y. & Fu, H.-G. Si-embedded graphene: an efficient and metal-free catalyst for CO oxidation by N2O or O2 . Theor Chem Acc. 131, 1242 (2012).
Article Google Scholar
Nigam, S. & Majumder, C. CO Oxidation by BN−Fullerene Cage: Effect of Impurity on the Chemical Reactivity. ACS Nano. 2, 1422–1428 (2008).
CASArticle Google Scholar
Sinthika, S., Kumar, E. M. & Thapa, R. Doped h-BN monolayer as efficient noble metal-free catalysts for CO oxidation: the role of dopant and water in activity and catalytic de-poisoning. J. Mater. Chem. A. 2, 12812–12820 (2014).
CASArticle Google Scholar
Silva, S. W. D., Du, A., Senadeera, W. & Gu, Y. Neutral and charged boron-doped fullerenes for CO2 adsorption. Beilstein J. Nanotechnol. 5, 413–418 (2014).
Article Google Scholar
Mahdavifar, Z., Abbasi, N. & Shakerzadeh, E. A comparative theoretical study of CO2 sensing using inorganic AlN, BN and SiC single walled nanotubes. Sensors and Actuators B. 185, 512–522 (2013).
CASArticle Google Scholar
Rana, M. K., Koh, H. S., Hwang, J. & Siegel, D. J. Comparing van der Waals Density Functionals for CO2 Adsorption in Metal Organic Frameworks. J. Phys. Chem. C. 116, 16957−16968 (2012).
CASArticle Google Scholar
Huang, B. et al. Adsorption of Gas Molecules on Graphene Nanoribbons and Its Implication for Nanoscale Molecule Sensor. J. Phys. Chem. C. 112, 13442–13446 (2008).
CASArticle Google Scholar
Sun, Q. et al. Charge-Controlled Switchable CO2 Capture on Boron Nitride Nanomaterials. J. Am. Chem. Soc. 135, 8246−8253 (2013).
CASArticle Google Scholar
Gao, B., Zhao, J.-X., Cai, Q.-H., Wang, X.-G. & Wang, X.-Z. Doping of Calcium in C60 Fullerene for Enhancing CO2 Capture and N2O Transformation: A Theoretical Study. J. Phys. Chem. A. 115, 9969–9976 (2011).
CASArticle Google Scholar
Shao, P., Kuang, X.-Y., Dinga, L.-P., Yang, J. & Zhong, M.-M. Can CO2 molecule adsorb effectively on Al-doped boron nitride single walled nanotube? Appl. Sur. Sci. 285, 350–356 (2013).
CASADSArticle Google Scholar
Gao, Y. et al. Nitrogen-Doped sp2-Hybridized Carbon as a Superior Catalyst for Selective Oxidation. Angew. Chem. Int. Ed. 52, 2109–2113 (2013).
CASArticle Google Scholar
Pramanik, A. & Kang, H. S. Density Functional Theory Study of O2 and NO Adsorption on Heteroatom-Doped Graphenes Including the van der Waals Interaction. J. Phys. Chem. C. 115, 10971–10978 (2011).
CASArticle Google Scholar
Wang, S.-G. et al. Factors Controlling the Interaction of CO2 with Transition Metal Surfaces. J. Phys. Chem. C. 111, 16934–16940 (2007).
CASArticle Google Scholar
Zhang, P. et al. Curvature effect of SiC nanotubes and sheets for CO2 capture and reduction. RSC Adv. 4, 48994–48999 (2014).
CASArticle Google Scholar
Cazorla, C., Shevlin, S. A. & Guo, Z. X. Calcium-Based Functionalization of Carbon Materials for CO2 Capture: A First-Principles Computational Study. J. Phys. Chem. C. 115, 10990–10995 (2011).
CASArticle Google Scholar
Atkins, P. & Paula, J. Atkins’ Physical Chemistry, 8th edn., (ed. W. H. Freeman and Company, New York, pp. 993–1001, 2006).
Cramer C. J Essentials of Computational Chemistry Theories and Models, 2nd ed. (John Wiley & Sons, pp. 355–365, 2005).
Duan, Z. & Henkelman, G. CO Oxidation on the Pd(111) Surface. ACS Catal. 4, 3435–3443 (2014).
CASArticle Google Scholar
Li, Y. et al. Stone−Wales Defects in Single-Walled Boron Nitride Nanotubes: Formation Energies, Electronic Structures and Reactivity. J. Phys. Chem. C. 112, 1365–1370 (2008).
CASArticle Google Scholar
Miyamoto, Y., Rubio, A., Berber, S., Yoon, M. & Tománek, D. Spectroscopic characterization of Stone-Wales defects in nanotubes. Phys. Rev. B 69, 121413 (2004).
ADSArticle Google Scholar
Gibb, A. L. et al. Atomic Resolution Imaging of Grain Boundary Defects in Monolayer Chemical Vapor Deposition-Grown Hexagonal Boron Nitride, J. Am. Chem. Soc. 135, 6758−6761 (2013).
CASArticle Google Scholar
An, W., Wu, X., Yang, J. L. & Zeng, X. C. Adsorption and Surface Reactivity on Single-Walled Boron Nitride Nanotubes Containing Stone-Wales Defects, J. Phys. Chem. C. 111, 14105–14112 (2007).
CASArticle Google Scholar
Gokhale, A. A., Dumesic, J. A. & Mavrikakis, M. On the Mechanism of Low-Temperature Water Gas Shift Reaction on Copper. J. Am. Chem. Soc. 130, 1402 – 1414 (2008).
CASArticle Google Scholar
Nørskov, J. K. et al. Origin of the Overpotential for Oxygen Reduction at a Fuel-Cell Cathode. J. Phys. Chem. B. 108, 17886–17892 (2004).
Article Google Scholar
Kresse, G. & Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mat. Sci. 6, 15–50 (1996).
CASArticle Google Scholar
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 77, 3865−3868 (1996).
CASADSArticle Google Scholar
Blöchl, P. E. Projector augmented-wave method. Phys. Rev. B: Condens. Matter Mater. Phys. 50, 17953–17979 (1994).
ADSArticle Google Scholar
Grimme, S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comp. Chem. 27, 1787 (2006).
CASArticle Google Scholar
Baroni, S., Giannozzi, P. & Testa, A. Green’s-function approach to linear response in solids. Phys. Rev. Lett. 58, 1861 (1987).
CASADSArticle Google Scholar
Jónsson, H., Mills, G. & Jacobsen, K. W. Nudged Elastic Band Method for Finding Minimum Energy Paths of Transitions, in Classical and Quantum Dynamics in Condensed Phase Simulations. 385, (Ed. B. J. Berne, G. Ciccotti & D. F. Coker, World Scientific, 1998).
Download references
Acknowledgements
RT and SS thank Science and Engineering Research Board (SERB), India for the financial support (Grant no: SB/FTP/PS028/2013). RT thanks SRM Research Institute, SRM University for providing supercomputing facility and financial support. Author KI would like to express their sincere thanks to the crew of Center for Computational Materials Science of the Institute for Materials Research, Tohoku University for their continuous support of the SR16000 supercomputing facilities. One of the authors (Y. K.) thanks the Russian Megagrant Project No.14.B25.31.0030 “New energy technologies and energy carriers” for supporting the present research. NP acknowledges the financial support of the Korea Institute of Science and Technology (Grant No. 2E25372).
Author information
Sinthika S. and Kumar E. Mathan contributed equally to this work.
Authors and Affiliations
SRM Research Institute, SRM University, Kattankulathur, 603203, Tamil Nadu, India
S. Sinthika, E. Mathan Kumar & Ranjit Thapa
New Industry Creation Hatchery Center (NICHe), Tohoku University, Sendai, Japan
V. J. Surya & Y. Kawazoe
Thermophysics Institute, Siberian Branch, Russian Academy of Sciences, Russia
Y. Kawazoe
Center for Multidimensional Carbon Materials, Institute for Basic Science (IBS), Ulsan, 689-798, Republic of Korea
Noejung Park
Department of Physics and Nanotechnology, SRM University, Kattankulathur, 603203
K. Iyakutti & Ranjit Thapa
Contributions
R.T. conceived the project and designed the problem. E.M.K. and K.I performed the stability calculation and S.S. performed the calculation for adsorption studies and catalytic reaction part. R.T., S.S. and E.M.K. made the figures. R.T., S.S. and E.M.K. wrote the manuscript. N.P. helped in writing the manuscript. K.I, V.J.S., Y.K. and N.P. helped to analyse the data. All authors reviewed the manuscript.
Lylia Hamidatou
- Department of Neutron Activation Analysis, Nuclear Research Centre of Birine, Algeria
Hocine Slamene
- Department of Neutron Activation Analysis, Nuclear Research Centre of Birine, Algeria
Tarik Akhal
- Department of Neutron Activation Analysis, Nuclear Research Centre of Birine, Algeria
Boussaad Zouranen
- Department of Neutron Activation Analysis, Nuclear Research Centre of Birine, Algeria
*Address all correspondence to:
1. Introduction
Analytical science to develop the methodology for the investigation of properties and structure of matter at level of single nucleus, atom and molecule, and scientific analysis to determine either chemical composition or elemental contents in a sample are indispensable in basic research and development, as well as in industrial applications.
Following the discovery of neutron by J. Chadwick in 1932 (Nobel prize, 1935) and the results of F. Joliot and I. Curie in 1934, neutron activation analysis was first developed by G. Hevesy and H. Levi in 1936. They used a neutron source (226Ra + Be) and a radiation detector (ionization chamber) and promptly recognized that the element Dy (dysprosium) in the sample became highly radioactive after exposure to the neutron source. They showed that the nuclear reaction may be used to determine the elements present in unknown samples by measuring the induced radioactivity.
Thereafter, the development of the nuclear reactors in the 1940s, the application of radiochemical techniques using low resolution scintillation detectors like NaI (Tl) in the 1950s, the development of semiconductor detectors (Ge, Si, etc.) and multichannel analyzer in the 1960s, and the advent of computers and relevant software in the 1970s, the nuclear technique has advanced to become an important analytical tool for determination of many elements at trace level. In spite of the developments in other chemical techniques, the simplicity and selectivity, the speed of operation, the sensitivity and accuracy of NAA have become and maintained its role as a powerful analytical technique. In 2011, Peter Bode describes in his paper “Neutron activation analysis: A primary method of measurements”, the history of the development of NAA overall the world [1].
Nowadays, there are many elemental analysis methods that use chemical, physical and nuclear characteristics. However, a particular method may be favoured for a specific task, depending on the purpose. Neutron activation analysis (NAA) is very useful as sensitive analytical technique for performing both qualitative and quantitative multielemental analysis of major, minor and traces components in variety of terrestrial samples and extra-terrestrial materials. In addition, because of its accuracy and reliability, NAA is generally recognized as the "referee method" of choice when new procedures are being developed or when other methods yield results that do not agree. It is usually used as an important reference for other analysis methods. Worldwide application of NAA is so widespread it is estimated that approximately 100,000 samples undergo analysis each year.
The method is based on conversion of stable atomic nuclei into radioactive nuclei by irradiation with neutrons and subsequent detection of the radiation emitted by the radioactive nuclei and its identification. The basic essentials required to carry out an analysis of samples by NAA are a source of neutrons, instrumentation suitable for detecting gamma rays, and a detailed knowledge of the reactions that occur when neutrons interact with target nuclei. Brief descriptions of the NAA method, reactor neutron sources, and gamma-ray detection are given below.
This chapter describes in the first part the basic essentials of the neutron activation analysis such as the principles of the NAA method with reference to neutron induced reactions, neutron capture cross-sections, production and decay of radioactive isotopes, and nuclear decay and the detection of radiation. In the second part we illustrated the equipment requirements neutron sources followed by a brief description of Es-Salam research reactor, gamma-ray detectors, and multi-channel analysers. In addition, the preparation of samples for neutron irradiation, the instrumental neutron activation analysis techniques, calculations, and systematic errors are given below. Some schemes of irradiation facilities, equipment and materials are given as examples in this section.
Finally, a great attention will be directed towards the most recent applications of the INAA and k0-NAA techniques applied in our laboratory. Examples of such samples, within a selected group of disciplines are milk, milk formulae and salt (nutrition), human hair and medicinal seeds (biomedicine), cigarette tobacco (environmental and health related fields) and iron ores (exploration and mining).
All steps of work were performed using NAA facilities while starting with the preparation of samples in the laboratory. The activation of samples depends of neutron fluence rate in irradiation channels of the Algerian Es-Salam research reactor. The radioactivity induced is measured by gamma spectrometers consist of germanium based semiconductor detectors connected to a computer used as a multichannel analyser for spectra evaluation and calculation. Sustainable developments of advanced equipment, facilities and manpower have been implemented to establish a state of the art measurement capability, to implement several applications, etc.
2. Neutron activation analysis
Neutron activation analysis (NAA) is a nuclear process used for determining the concentrations of elements in a vast amount of materials. NAA relies on excitation by neutrons so that the treated sample emits gamma-rays. It allows the precise identification and quantification of the elements, above all of the trace elements in the sample. NAA has applications in chemistry but also in other research fields, such as geology, archaeology, medicine, environmental monitoring and even in the forensic science.
2.1. Basis principles
The sequence of events occurring during the most common type of nuclear reaction used for NAA, namely the neutron capture or (n, gamma) reaction, is illustrated in Figure 1. Creation of a compound nucleus forms in an excited state when a neutron interacts with the target nucleus via a non-elastic collision. The excitation energy of the compound nucleus is due to the binding energy of the neutron with the nucleus. The compound nucleus will almost instantaneously de-excite into a more stable configuration through emission of one or more characteristic prompt gamma rays. In many cases, this new configuration yields a radioactive nucleus which also de-excites (or decays) by emission of one or more characteristic delayed gamma rays, but at a much lower rate according to the unique half-life of the radioactive nucleus. Depending upon the particular radioactive species, half-lives can range from fractions of a second to several years.
In principle, therefore, with respect to the time of measurement, NAA falls into two categories: (1) prompt gamma-ray neutron activation analysis (PGNAA), where measurements take place during irradiation, or (2) delayed gamma-ray neutron activation analysis (DGNAA), where the measurements follow radioactive decay. The latter operational mode is more common; thus, when one mentions NAA it is generally assumed that measurement of the delayed gamma rays is intended. About 70% of the elements have properties suitable for measurement by NAA.
The PGAA technique is generally performed by using a beam of neutrons extracted through a reactor beam port. Fluxes on samples irradiated in beams are in the order of one million times lower than on samples inside a reactor but detectors can be placed very close to the sample compensating for much of the loss in sensitivity due to flux. The PGAA technique is most applicable to elements with extremely high neutron capture cross-sections (B, Cd, Sm, and Gd); elements which decay too rapidly to be measured by DGAA; elements that produce only stable isotopes (e.g. light elements); or elements with weak decay gamma-ray intensities. 2D, 3D-analysis of (main) elements distribution in the samples can be performed by PGAA.
DGNAA (sometimes called conventional NAA) is useful for the vast majority of elements that produce radioactive nuclides. The technique is flexible with respect to time such that the sensitivity for a long-lived radionuclide that suffers from interference by a shorter-lived radionuclide can be improved by waiting for the short-lived radionuclide to decay or quite the contrary, the sensitivity for short-lived isotopes can be improved by reducing the time irradiation to minimize the interference of long-lived isotopes. This selectivity is a key advantage of DGNAA over other analytical methods.
In most cases, the radioactive isotopes decay and emit beta particles accompanied by gamma quanta of characteristic energies, and the radiation can be used both to identify and accurately quantify the elements of the sample. Subsequent to irradiation, the samples can be measured instrumentally by a high resolution semiconductor detector, or for better sensitivity, chemical separations can also be applied to reduce interferences. The qualitative characteristics are: the energy of the emitted gamma quanta (Eγ) and the half life of the nuclide (T½). The quantitative characteristic is: the Iγ intensity, which is the number of gamma quanta of energy Eγ measured per unit time.
The n-gamma reaction is the fundamental reaction for neutron activation analysis. For example, consider the following reaction:
58Fe is a stable isotope of iron while 59Fe is a radioactive isotope. The gamma rays emitted during the decay of the 59Fe nucleus have energies of 142.4, 1099.2, and 1291.6 KeV, and these gamma ray energies are characteristic for this nuclide (see figure 2) [2]. The probability of a neutron interacting with a nucleus is a function of the neutron energy. This probability is referred to as the capture cross-section, and each nuclide has its own neutron energy-capture cross-section relationship. For many nuclides, the capture cross-section is greatest for low energy neutrons (referred to as thermal neutrons). Some nuclides have greater capture cross-sections for higher energy neutrons (epithermal neutrons). For routine neutron activation analysis we are generally looking at nuclides that are activated by thermal neutrons.
The most common reaction occurring in NAA is the (n,γ) reaction, but also reactions such as (n,p), (n,α), (n,n′) and (n,2n) are important. The neutron cross section, σ, is a measure for the probability that a reaction will take place, and can be strongly different for different reaction types, elements and energy distributions of the bombarding neutrons. Some nuclei, like 235U are fissionable by neutron capture and the reaction is denoted as (n,f), yielding fission products and fast (highly energetic) neutrons [1].
Neutrons are produced via
Isotopic neutron sources, like 226Ra(Be), 124Sb(Be), 241Am(Be), 252Cf. The neutrons have different energy distributions with a maximum in the order of 3–4 MeV; the total output is typically 105–107 s -1 GBq-1 or, for 252Cf, 2.2 1012 s-1g-1.
Particle accelerators or neutron generators. The most common types are based on the acceleration of deuterium ions towards a target containing either deuterium or tritium, resulting in the reactions 2H(2H,n)3He and 3H(2H,n)4He, respectively. The first reaction, often denoted as (D,D), yields monoenergetic neutrons of 2.5 MeV and typical outputs in the order of 108–1010 s−1; the second reaction (D,T) results in monoenergetic neutrons of 14.7 MeV and outputs of 109–1011 s−1.
Nuclear research reactors. The neutron energy distribution depends on design of the reactor and its irradiation facilities. An example of an energy distribution in a light water moderated reactor is given in Fig. 2.3 from which it can be seen that the major part of the neutrons has a much lower energy distribution that in isotopic sources and neutron generators. The neutron output of research reactors is often quoted as neutron fluence rate in an irradiation facility and varies, depending on reactor design and reactor power, between 1015 and 1018 m-2 s-1.
Owing to the high neutron flux, experimental nuclear reactors operating in the maximum thermal power region of 100 kW -10 MW with a maximum thermal neutron flux of 1012-1014 neutrons cm-2 s-1 are the most efficient neutron sources for high sensitivity activation analysis induced by epithermal and thermal neutrons. The reason for the high sensitivity is that the cross section of neutron activation is high in the thermal region for the majority of the elements. There is a wide distribution of neutron energy in a reactor and, therefore, interfering reactions must be considered. In order to take these reactions into account, the neutron spectrum in the channels of irradiation should be known exactly. E.g. if thermal neutron irradiations are required, the most thermalized channels should be chosen.
Although there are several types of neutron sources (reactors, accelerators, and radioisotopic neutron emitters) one can use for NAA, nuclear reactors with their high fluxes of neutrons from uranium fission offer the highest available sensitivities for most elements. Different types of reactors and different positions within a reactor can vary considerably with regard to their neutron energy distributions and fluxes due to the materials used to moderate (or reduce the energies of) the primary fission neutrons. This is further elaborated in the title “Derivation of the measurement equation”. In our case, the NAA method is based on the use of neutron flux in several irradiation channels of Es-Salam Research reactor. In 2011, Hamidatou L et Al., reported “Experimental and MCNP calculations of neutron flux parameters in irradiation channel at Es-Salam reactor” the core modelling to calculate neutron spectra using experimental and MCNP approaches. The Es-Salam reactor was designed for a thermal power output of 15 Mw, with 72 cylindrical cluster fuel elements; each fuel element consists of 12 cylindrical rods of low enriched UO2. In addition the both of fuel throttle tube of the cluster and fuel element tube encloses heavy water as moderator and coolant. The fuel elements are arranged on a heavy water square lattice. The core of the reactor is constituted by a grid containing 72 fuel elements, 12 rods for reactivity control and two experimental channels.
There is also a heavy water in the middle of the core including five experimental channels called inner reflector, In addition, all fuel elements have a reflector at each end called upper and lower reflector. The core is reflected laterally by heavy water maintained in aluminium tank followed by the graphite.
2.2. Neutron activation analysis procedure
In the majority of INAA procedures thermal reactor neutrons are used for the activation: neutrons in thermal equilibrium with their environment. Sometimes activation with epithermal reactor neutrons (neutrons in the process of slowing down after their formation from fission of 235U) is preferred to enhance the activation of elements with a high ratio of resonance neutron cross section over thermal neutron cross section relatively to the activation of elements with a lower such a ratio. In principle materials can be activated in any physical state, viz. solid, liquid or gaseous. There is no fundamental necessity to convert solid material into a solution prior to activation; INAA is essentially considered to be a non-destructive method although under certain conditions some material damage may occur due to thermal heating, radiolysis and radiation tracks by e.g. fission fragments and α-radiation emitting nuclei. It is essential to have more than two or three qualified full-time member of the staff with responsibility for the NAA facilities. They should be able to control the counting equipment and have good knowledge of basic principles of the technique. In addition, the facility users and the operators must establish a good channel of communication. Other support staff will be required to maintain and improve the equipment and facility. It seems, therefore, a multi-disciplinary team could run the NAA system well.
The analytical procedure is based on four steps:
sample preparation (Figure 3) means in most cases only heating or freeze drying, crushing or pulverization, fractionating or pelletizing, evaporation or pre-concentration, put through a sieve, homogenising, weighing, washing, check of impurities (blank test), encapsulation and sealing irradiation vial, as well as the selection of the best analytical process and the preparation of the standards. The laboratory ambiance is also important for preservation and storage of the samples. Standardization is the basis for good accuracy of analytical tools and often depends on particular technology, facility and personnel. For production of accurate data, careful attention to all possible errors in preparing single or multi-element standards is important, and standards must be well chosen depending on the nature of the samples.
irradiation of samples can be taken from the various types of neutron sources according to need and availability. For the INAA, one pneumatic transfer system installed in the horizontal channel at Es-Salam research reactor for short irradiation of samples (Figure 4). In addition, two vertical channels located in different sites of the heavy water moderator and the graphite reflector have been used for long irradiations. The neutron spectrum parameters at different irradiation channels such as alpha, f, Tn, etc are experimentally determined using cadmium ratio, cadmium cover, bare triple monitor and bi-isotopic methods using HΦgdhal convention and Westcott formalism Table 1 and Table 2. The calibration of the irradiation positions has been carried out to implement the k0-NAA in our laboratory.
| Cd-ratio | 0.026±0.012 | 28.4±1.6 | 0.038±0.004 |
| Cd-covered | 0.024±0.010 | 28.7±2.1 | - |
| Bare triple monitor | 0.030±0.008 | 28.6±1.8 | - |
| Bare bi-isotopic | - | 29.5±2.5 | 0.036±0.003 |
| Average | 0.027±0.010 | 28.8±2.0 | 0.037±0.003 |
Table 1.
The parameters α, f and obtained by different methods.
| Measured value | 0.027±0.010 | 28.8±2.0 | 34±1.8 | 2.93±0.32 | 0.037±0.003 |
Table 2.
Neutron spectrum parameters in the irradiation site at es-Salam research reactor.
after the irradiation the measurement is performed after a suitable cooling time (tc). In NAA, nearly exclusively the (energy of the) gamma radiation is measured because of its higher penetrating power of this type of radiation, and the selectivity that can be obtained from distinct energies of the photons - differently from beta radiation which is a continuous energy distribution. The interaction of gamma- and X-radiation with matter results, among others, in ionization processes and subsequent generation of electrical signals (currents) that can be detected and recorded.
The instrumentation used to measure gamma rays from radioactive samples generally consists of a semiconductor detector, associated electronics, and a computer-based multi-channel analyzer (MCA/computer).
Most NAA labs operate one or more hyper-pure germanium (HPGe) detectors, which operate at liquid nitrogen temperature (77 K). Although HPGe detectors come in many different shapes and sizes, the most common shape is coaxial. These detectors are very useful for measurement of gamma rays with energies in the range from about 60 keV to 3.0 MeV. The two most important characteristics a HPGe detector are its resolution and efficiency. Other characteristics to consider are peak shape, peak-to-Compton ratio, pulse rise time, crystal dimensions or shape, and price. The detector’s resolution is a measure of its ability to separate closely spaced peaks in the spectrum, and, in general, the resolution is specified in terms of the full width at half maximum (FWHM) of the 122 keV photopeak of 57Co and the 1,332 keV photopeak of 60Co. For most NAA applications, a detector with 0.5 keV resolution or less at 122 keV and 1.8 keV or less at 1,332 keV is sufficient. Detector efficiency for a given detector depends on gamma-ray energy and the sample and detector geometry, i.e. subtended solid angle. Of course, a larger volume detector will have a higher efficiency.
At Es-Salam NAA Lab, four gamma-ray spectrometers of Canberra for which one of them consists of a HPGe detector 35% relative efficiency connected with Genie 2k Inspector and the three other spectrometers are composed of detectors (30, 35 and 45 % relative efficiency) connected with a three Lynx® Digital Signal Analyser, It is a 32K channel integrated signal analyzer based on advanced digital signal processing (DSP) techniques. All spectrometers operate with Genie™2000 spectroscopy software. A radiation detector therefore consists of an absorbing material in which at least part of the radiation energy is converted into detectable products, and a system for the detection of these products. Figure 5 illustrates Gamma-ray spectroscopy systems. The detectors are kept at liquid nitrogen temperatures (dewers under cave). The boxes in the left and in the right of the computer are the Lynx Digital Spectrometer Processing.
Measurement, evaluation and calculation involve taking the gamma spectra and the calculating trace element concentrations of the sample and preparation of the NAA report.
In this part of work, Peter bode describes clearly in his paper [1] the analysis procedure of gamma-spectrum to the determination of the amount of element in sample. The acquisition of gamma spectrum Fig.6 and Fig.7 via the spectroscopy system Fig. 5 is analyzed to identify the radionuclides produced and their amounts of radioactivity in order to derive the target elements from which they have been produced and their masses in the activated sample. The spectrum analysis starts with the determination of the location of the (centroids of the) peaks. Secondly, the peaks are fitted to obtain their precise positions and net peak areas. The Analytical protocol adopted in our NAA laboratory is presented in Fig.8.
The positions – often expressed as channel numbers of the memory of a multi-channel pulse height analyzer – can be converted into the energies of the radiation emitted; this is the basis for the identification of the radioactive nuclei. On basis of knowledge of possible nuclear reactions upon neutron activation, the (stable) element composition is derived. The values of the net peak areas can be used to calculate the amounts of radioactivity of the radionuclides using the full energy photopeak efficiency of the detector.
The amounts (mass) of the elements may then be determined if the neutron fluence rate and cross sections are known. In the practice, however, the masses of the elements are determined from the net peak areas by comparison with the induced radioactivity of the same neutron activation produced radionuclides from known amounts of the element of interest. The combination of energy of emitted radiation, relative intensities if photons of different energies are emitted and the half life of the radionuclide is unique for each radionuclide, and forms the basis of the qualitative information in NAA. The amount of the radiation is directly proportional to the number of radioactive nuclei produced (and decaying), and thus with the number of nuclei of the stable isotope that underwent the nuclear reaction. It provides the quantitative information in NAA.
The measured in NAA – the quantity intended to be measured – is the total mass of a given element in a test portion of a sample of a given matrix in all physico-chemical states. The quantity ‘subject to measurement’ is the number of disintegrating nuclei of a radionuclide. The measurement results in the number of counts in a given period of time, from which the disintegration rate and the number of disintegrating nuclei is calculated; the latter number is directly proportional to the number of nuclei of the stable isotope subject to the nuclear reaction, and thus to the number of nuclei of the element, which finally provides information on the mass and amount of substance of that element (see Eq. 16). An example of typical ranges of experimental conditions is given in Table 3 [1].
In practice, our laboratory proceeds in the treatment of spectra and calculation of elemental concentrations of analyzed samples according the approach illustrated in figure 8.
| Test portion mass : 5-500 mg | |||
| Neutron fluence rates available 1016 – 1018 m-2 s-1 | |||
| Irradiation | Decay | Measurement | Analyzed element |
| 5 – 30 seconds | 5 – 600 seconds | 15 – 300 seconds | Short lived |
| 1 – 8 hours | 3 – 5 days | 1 – 4 hours | Medium lived |
| 20 days | 1 – 16 hours | Long lived | |
Table 3.
Example of typical ranges of experimental conditions of an INAA procedure.
2.3. Derivation of the measurement equation
The reaction rate R per nucleus capturing a neutron is given by:
E1
where:
σ (v) is the (n,γ) cross section (in cm2 ; 1 barn (b) = 10-24 cm2) at neutron velocity v (in cm s-1);
σ (E) is the (n,γ) cross section (in cm2) at neutron energy E (in eV);
Φ’(v) is the neutron flux per unit of velocity interval (in cm-3) at neutron velocity v;
n’(v) is the neutron density per unit of velocity interval (in cm-4 s) at neutron velocity v;
Φ’(E) is the neutron flux per unit of energy interval (in cm-2 s-1 eV-1) at neutron energy E.
In Eq.(1), σ (v) = σ (E) with E (in erg = 6.2415.1011 eV) = ½ mn v2 [mn rest mass of the neutron = 1.6749 10-24 g]. Furthermore, per definition, φ’(v) dv = φ’(E)dE (both in cm-2 s-1).
In Eq.1, the functions σ(v) [= σ (E)] and φ’(v) [ φ’(E)] are complex and are respectively depending on the (n,γ) reaction and on the irradiation site.
In 1987, F De Corte describes in his Aggregate thesis “Chapter 1: fundamentals [3] that the introduction of some generally valid characteristics yields the possibility of avoiding the actual integration and describing accurately the reaction rate in a relatively simple way by means of so-called formalisms or conventions. In short, these characteristics are:
In nuclear research reactors – which are intense sources of neutrons – three types of neutrons can be distinguished. The neutron flux distribution can be divided into three components (see Figure 9):
Fission or fast neutrons released in the fission of 235U. Their energy distribution ranges from 100 keV to 25 MeV with a maximum fraction at 2 MeV. These neutrons are slowed down by interaction with a moderator, e.g. H2O, to enhance the probability of them causing a fission chain reaction in the 235U.
The epithermal neutron component consists of neutrons (energies from 0.5 eV to about 100 keV). A cadmium foil 1 mm thick absorbs all thermal neutrons but will allow epithermal and fast neutrons above 0.5 eV in energy to pass through. Both thermal and epithermal neutrons induce (n,γ) reactions on target nuclei.
The thermal neutron component consists of low-energy neutrons (energies below 0.5 eV) in thermal equilibrium with atoms in the reactor's moderator. At room temperature, the energy spectrum of thermal neutrons is best described by a Maxwell-Boltzmann distribution with a mean energy of 0.025 eV and a most probable velocity of 2200 m/s. In general, a 1 MW reactor has a peak thermal neutron flux of approximately 1013 n/cm2.
The (n,γ) cross section function, σ(v) versus v can be interpreted as a σ(v) ~ 1/v dependence, or σ (E) ~ 1/E1/2 dependence [log σ (E) versus log E is linear with slope -1/2], on which (above some eV) several resonances are superposed see Figure 10 taken from http://thorea.wikia.com/wiki/Thermal,_Epithermal_and_Fast_Neutron_Spectra web page.
An NAA technique that employs only epithermal neutrons to induce (n,γ) reactions by irradiating the samples being analyzed inside either cadmium or boron a shield is called epithermal neutron activation analysis (ENAA).
The production of radioactive nuclei is described by:
E2
In which N0 number of target nuclei, N is the number of radioactive nuclei, λ is the decay constant in s−1. The disintegration rate of the produced radionuclide at the end of the irradiation time ti follows from:
E3
where:
D is the disintegration rate in Bq of the produced radionuclide, assuming that N=0 at t=0 and N0=constant.
The dependence of the activation cross section and neutron fluence rate to the neutron energy can be taken into account in Eq. (1) by dividing the neutron spectrum into a thermal and an epithermal region; the division is made at En=0.55 eV (the so-called cadmium cut-off energy). This approach is commonly known as the Høgdahl convention [4].
The integral in Eq. (1) can then be rewritten as:
E4
The first term can be integrated straightforward:
E5
in which,
E6
is called the thermal neutron density, with Φth=nv0,
Φth is the conventional thermal neutron fluence rate, m−2 s−1, for energies up to the Cd cut-off energy of 0.55 eV;
σ0 is the thermal neutron activation cross section, m2, at 0.025 eV;
v0 is the most probable neutron velocity at 20 °C: 2200 m s−1.
The second term is re-formulated in terms of neutron energy rather than neutron velocity and the infinite dilution resonance integral I0 – which effectively is also a cross section (m2) – is introduced:
E7
with:
E8
Here, Φepi the conventional epithermal neutron fluence rate per unit energy interval, at 1 eV.
From this definition of I0 it can be seen that it assumes that the energy dependency of the epithermal neutron fluence rate is proportional to 1/En. This requirement is fulfilled to a good approximation by most of the (n,γ) reactions.
In the practice of nuclear reactor facilities the epithermal neutron fluence rate Φepi is not precisely following the inverse proportionality to the neutron energy; the small deviation can be accounted for by introducing an epithermal fluence rate distribution parameter α:
E9
The expression for the reaction rate can thus be re-written as:
E10
Expressing the ratio of the thermal neutron fluence rate and the epithermal neutron fluence rate as f=Φth/Φepi and the ratio of the resonance integral and the thermal activation cross section as Q0(α)= I0(α)/σ0, an effective cross section can be defined:
E11
It simplifies the Eq. (10) for the reaction rate to:
E12
This reaction rate applies to infinite thin objects. In objects of defined dimensions, the inside part will experience a lower neutron fluence rate than the outside part because neutrons are removed by absorption.
The nuclear transformations are established by measurement of the number of nuclear decays. The number of activated nuclei N(ti,td) present at the start of the measurement is given by:
E13
and the number of nuclei ΔN disintegrating during the measurement is given by:
E14
in which td is the decay or waiting time, i.e. the time between the end of the irradiation and the start of the measurement tm is the duration of the measurement. Additional correction resulting from high counting rates may be necessary depending upon the gamma-ray spectrometer hardware used as illustrated in chapter 2 [1]. Replacing the number of target nuclei N0 by (NAvm)/M and using the Eq. (12) for the reaction rate, the resulting net counts C in a peak in the spectrum corresponding with a given photon energy is approximated by the activation formula:
E15
with:
Np is the net counts in the γ-ray peak of Eγ ;
NAv is the Avogadro's number in mol−1;
θ is isotopic abundance of the target isotope;
mx is the mass of the irradiated element in g;
Ma is the atomic mass in g mol−1;
I is the gamma-ray abundance, i.e. the probability of the disintegrating nucleus emitting a photon of Eγ (photons disintegration−1);
ε is the full energy photopeak efficiency of the detector, i.e. the probability that an emitted photon of given energy will be detected and contribute to the photopeak at energy Eγ in the spectrum.
Although the photons emitted have energies ranging from tens of keVs to MeVs and have high penetrating powers, they still can be absorbed or scattered in the sample itself depending on the sample size, composition and photon energy. This effect is called gamma-ray self-attenuation. Also, two or more photons may be detected simultaneously within the time resolution of the detector; this effect is called summation.
Eq. (15) can be simply rewritten towards the measurement equation of NAA, which shows how the mass of an element measured can be derived from the net peak area C:
E16
2.4. Standardization
Standardization is based on the determination of the proportionality factors F that relate the net peak areas in the gamma-ray spectrum to the amounts of the elements present in the sample under given experimental conditions:
E17
Both absolute and relative methods of calibration exist.
2.4.1.Absolute calibration
The values of the physical parameters determining the proportionality factor θ, NAv, M, σeff I, λ, are taken from literature. The parameters σeff respectively I, λ are not precisely known for many (n,γ) reactions and radionuclides, and in some cases θ is also not accurately known. Since the various parameters were often achieved via independent methods, their individual uncertainties will add up in the combined uncertainty of measurement of the elemental amounts, leading to a relatively large combined standard uncertainty. Moreover, the metrological traceability of the values of the physical constants is not known for all radionuclides. The other parameters Np, mx, Φ, ε, ti, td, tm are determined, calculated or measured for the given circumstances and uncertainties can be established.
2.4.2. Relative calibration
Direct comparator method
The unknown sample is irradiated together with a calibrator containing a known amount of the element(s) of interest. The calibrator is measured under the same conditions as the sample (sample-to-detector distance, equivalent sample size and if possible equivalent in composition). From comparison of the net peak areas in the two measured spectra the mass of the element of interest can be calculated:
E18
in which mx(unk), mx(cal) mass of the element of interest, in the unknown sample and the calibrator, respectively in g.
In this procedure many of the experimental parameters - such as neutron fluence rate, cross section and photopeak efficiency cancel out at the calculation of the mass and the remaining parameters are all known. This calibration procedure is used if the highest degree of accuracy is required.
The relative calibration on basis of element calibrators is not immediately suitable for laboratories aiming at the full multi-element powers of INAA. It takes considerable effort to prepare multi-element calibrators for all 70 elements measurable via NAA with adequate degree of accuracy in a volume closely matching the size and the shape of the samples. Single comparator method Multi-element INAA on basis of the relative calibration method is feasible when performed according to the principles of the single comparator method. Assuming stability in time of all relevant experimental conditions, calibrators for all elements are co-irradiated each in turn with the chosen single comparator element. Once the sensitivity for all elements relative to the comparator element has been determined (expressed as the so-called k-factor, see below), only the comparator element has to be used in routine measurements instead of individual calibrators for each element. The single comparator method for multi-element INAA was based on the ratio of proportionality factors of the element of interest and of the comparator element after correction for saturation, decay, counting and sample weights defined the k-factor for each element i as:
E19
Masses for each element i then can be calculated from these ki factors; for an element determined via a directly produced radionuclide the mass mx(unk) follows from:
E20
where: mx(comp) is the mass of element x in comparator in g.
These experimentally determined k-factors are often more accurate than when calculated on basis of literature data as in the absolute calibration method. However, the k-factors are only valid for a specific detector, a specific counting geometry and irradiation facility, and remain valid only as long as the neutron fluence rate parameters of the irradiation facility remain stable. The single comparator method requires laborious calibrations in advance, and finally yield relatively (compared to the direct comparator method) higher uncertainties of the measured values. Moreover, it requires experimental determination of the photopeak efficiencies of the detector. Metrological traceability of the measured values to the S.I. may be demonstrated.
The k0-comparator method
The 0-based neutron activation analysis (0-NAA) technique, developed in 1970s, is being increasingly used for multielement analysis in a variety of matrices using reactor neutrons [4-10]. In our research reactor, the 0-method was successfully developed using the Høgdahl formalism [11]. In the 0-based neutron activation analysis the evaluation of the analytical result is based on the so-called 0- factors that are associated with each gamma-line in the gamma-spectrum of the activated sample. These factors replace nuclear constants, such as cross sections and gamma-emission probabilities, and are determined in specialized NAA laboratories. This technique has been reported to be flexible with respect to changes in irradiation and measuring conditions, to be simpler than the relative comparator technique in terms of experiments but involves more complex formulae and calculations, and to eliminate the need for using multielement standards. The 0-NAA technique, in general, uses input parameters such as (1) the epithermal neutron flux shape factor (α), (2) subcadmium-to-epithermal neutron flux ratio (), (3) modified spectral index, (4) Westcott’s ()-factor, (5) the full energy peak detection efficiency (), and (6) nuclear data on 0 (ratio of resonance integral (0) to thermal neutron cross section (σ0) and 0. The parameters from (1) to (4) are dependent on each irradiation facility and the parameter (5) is dependent on each counting facility. The neutron field in a nuclear reactor contains an epithermal component that contributes to the sample neutron activation [12]. Furthermore, for nuclides with the Westcott’s ()-factor different from unity, the Høgdahl convention should not be applied and the neutron temperature should be introduced for application of a more sophisticated formalism [14], the Westcott formalism. These two formalisms should be taken into account in order to preserve the accuracy of 0-method.
The 0-NAA method is at present capable of tackling a large variety of analytical problems when it comes to the multi-element determination in many practical samples. In this part, we have published a paper [15] for which the determination of the Westcott and Høgdahl parameters have been carried out to assess the applicability of the 0-NAA method using the experimental system and irradiation channels at Es-Salam research reactor.
During the three last decades Frans de Corte and his co-workers focused their investigations to develop a method based on co-irradiation of a sample and a neutron flux monitor, such as gold and the use of a composite nuclear constant called k0-factor [3, 16]. In addition, this method allows to analyze the sample without use the reference standard like INAA method. The k-factors have been defined as independent of neutron fluence rate parameters as well as of spectrometer characteristics. In this approach, the irradiation parameter (1+Q0(α)/f) (Eq. (11)) and the detection efficiency ε are separated in the expression (19) of the k-factor, which resulted at the definition of the k0-factor.
E21
E22
The applicability of HØGDAHL convention is restricted to (n,γ) reactions for which WESTCOTT’s g-factor is equal to unity (independent of neutron temperature), the cases for which WESTCOTT’s g = 1 [3, 4, 17], such as the reactions 151Eu(n, γ) and 176Lu(n, γ) are excluded from being dealt with. Compared with relative method k0-NAA is experimentally simpler (it eliminates the need for multi-element standards [3, 18], but requires more complicated calculations [19]. In our research reactor, the k0-method was successfully developed using the HØGDAHL convention and WESTCOTT formalism [11, 15]. The k0-method requires tedious characterizations of the irradiation and measurement conditions and results, like the single comparator method, in relatively high uncertainties of the measured values of the masses. Moreover, metrological traceability of the currently existing k0 values and associated parameters to the S.I. is not yet transparent and most probably not possible. Summarizing, relative calibration by the direct comparator method renders the lowest uncertainties of the measured values whereas metrological traceability of these values to the S.I. can easily be demonstrated. As such, this approach is often preferred from a metrological viewpoint. The concentration of an element can be determined as:
E23
Where: the indices x and Au refer to the sample and the monitor, respectively; WAu and Wx represent the mass of the gold monitor and the sample (in g); Np is the measured peak area, corrected for dead time and true coincidence; S, D, C are the saturation, decay and counting factors, respectively; tm is the measuring time; Gth and Ge are the correction factors for thermal and epithermal neutron self shielding, respectively.
2.5. Sources errors
Many publications reported in literature [20-25] treat the concept of evaluation of uncertainties in large range of analytical techniques.
We can give in this part of chapter, the evaluation of uncertainties for neutron activation analysis measurements. Among the techniques of standardization the comparator method for which the individual uncertainty components associated with measurements made with neutron activation analysis (NAA) using the comparator method of standardization (calibration), as well as methods to evaluate each one of these uncertainty components [1].
This description assumes basic knowledge of the NAA method, and that experimental parameters including sample and standard masses, as well as activation, decay, and counting times have been optimized for each measurement. It also assumes that the neutron irradiation facilities and gamma-ray spectrometry systems have been characterized and optimized appropriately, and that the choice of irradiation facility and detection system is appropriate for the measurement performed. Careful and thoughtful experimental design is often the best means of reducing uncertainties. The comparator method involves irradiating and counting a known amount of each element under investigation using the same or very similar conditions as used for the unknown samples. Summarizing, relative calibration by the direct comparator method renders the lowest uncertainties of the measured values whereas metrological traceability of these values to the S.I. can easily be demonstrated. As such, this approach is often preferred from a metrological viewpoint. The measurement equation can be further simplified, by substituting:
E24
in :
E25
Where: Rθ is the ratio of isotopic abundances for unknown sample and calibrator, Rϕ is the ratio of neutron fluence rates (including fluence gradient, neutron self shielding, and scattering) for unknown sample and calibrator, Rσ is the ratio of effective cross sections if neutron spectrum shape differs from unknown sample to calibrator, Rε is the ratio of counting efficiencies (differences due to geometry and γ-ray self shielding) for unknown sample and calibrator, blank is the mass of element x in the blank, fP is the correction for pulse pileup (correction method depends upon the actual hardware used) and fltc is the correction for inadequacy of live time extension (correction method depends upon the actual hardware used)
Note that the R values are normally very close to unity, and all units are either SI-based or dimensionless ratios. Thus an uncertainty budget can be developed using only SI units and dimensionless ratios for an NAA measurement by evaluating the uncertainties for each of the terms in Eqs. (23) and (24), and for any additional corrections required (e.g., interferences, dry mass conversion factors, etc.).
Uncertainties for some of the terms in Eq. (24) have multiple components. If we sub-divide the uncertainty for each term in the above equations into individual components, add terms for potential corrections, and separate into the four stages of the measurement process, including: pre-irradiation (sample preparation); irradiation; post-irradiation (gamma-ray spectrometry), and radiochemistry, we arrive at the complete list of individual uncertainty components for NAA listed below in Table 4. Only uncertainties from the first three stages should be considered for instrumental neutron activation analysis (INAA) measurements, while all four stages should be considered for radiochemical neutron activation analysis (RNAA) measurements. More details are given in chapter 2 of reference [1] for each subsection of uncertainty component.
| 1. Pre-irradiation (sample and standard preparation) stage | |||
| 1.1. Elemental content of standards (comparators) | |||
| 1.2. Target isotope abundance ratio — unknown samples/standards | |||
| 1.3. Basis mass (or other sample basis) — including drying | |||
| 1.4. Sample and standard blanks | |||
| 2. Irradiation stage | |||
| 2.1. Neutron fluence exposure differences (ratios) for unknown samples compared to standards (comparators) | |||
| 2.1. Physical effects (fluence gradients within a single irradiation) | |||
| 2.2. Temporal effects (fluence variations with time) | |||
| 2.3. Neutron self shielding (absorption and scattering) effects within a single sample or standard | |||
| 2.4. Neutron shielding effects from neighbouring samples or standards | |||
| 2.2. Irradiation interferences | |||
| 2.2.1. Fast (high energy) neutron interferences | |||
| 2.2.2. Fission interferences | |||
| 2.2.3. Multiple neutron capture interferences | |||
| 2.3. Effective cross section differences between samples and standards | |||
| 2.4. Irradiation losses and gains | |||
| 2.4.1. Hot atom transfer (losses and gains by recoil, nanometer movement) | |||
| 2.4.2. Transfer of material through irradiation container | |||
| 2.4.3. Sample loss during transfer from irradiation container | |||
| 2.4.4. Target isotope burn up differences | |||
| 2.5. Irradiation timing and decay corrections during irradiation (effects of half life and timing uncertainties) | |||
| 3. Gamma-ray spectrometry stage | |||
| 3.1. Measurement replication or counting statistics (depending on number of replicates) for unknown samples | |||
| 3.2. Measurement replication or counting statistics (depending on number of replicates) for comparator standards | |||
| 3.3. Corrections for radioactive decay (effects of half life and timing uncertainties for each measurement) | |||
| 3.3.1. From end of irradiation to start of measurement | |||
| 3.3.2. Effects of clock time uncertainty | |||
| 3.3.3. Effects of live time uncertainty | |||
| 3.3.4. Count-rate effects for each measurement | |||
| 3.3.4.1. Corrections for losses due to pulse pileup for conventional analyzer systems | |||
| 3.3.4.2. Effects due to inadequacy of live-time extension for conventional analyzer systems | |||
| 3.3.4.3. Uncertainties due to hardware corrections for Loss-Free or Zero Dead Time systems | |||
| 3.4. Corrections for gamma-ray interferences | |||
| 3.5. Corrections for counting efficiency differences (if necessary), or uncertainty for potential differences | |||
| 3.5.1. Effects resulting from physical differences in size and shape of samples versus standards | |||
| 3.5.2. Corrections for gamma-ray self absorption | |||
| 3.6. Potential bias due to peak integration method | |||
| 3.7. Potential bias due to perturbed angular correlations (-ray directional effects) | |||
| 4. Radiochemical stage (only if radiochemical separations are employed) | |||
| 4.1. Losses during chemical separation | |||
| 4.2. Losses before equilibration with carrier or tracer | |||
Table 4.
Complete list of individual uncertainty components for NAA measurements using the comparator method of standardization; line numbers in this table represent subsections.
2.6. Detection limits of NAA
The detection limit represents the ability of a given NAA procedure to determine the minimum amounts of an element reliably. The detection limit depends on the irradiation, the decay and the counting conditions. It also depends on the interference situation including such things as the ambient background, the Compton continuum from higher energy-rays, as well as any-ray spectrum interferences from such factors as the blank from pre-irradiation treatment and from packing materials. The detection limit is often calculated using Currie's formula:
E26
where: DL is the detection limit and B is the background under a gamma-ray peak. This relation is valid only when the gamma-ray background (counting statistical error) is the major interference.
However, practically, the INAA detection limits depend on:
The amount of material to be irradiated and to be counted. This is often set by availability, sample encapsulation aspects and safety limits both related to irradiation (irradiation containers) and counting (e.g. with Ge well-type detectors), and possibly because of neutron self-shielding and gamma-ray self-absorption effects. For these reasons practically the sample mass is often limited to approximately 250 mg.
The neutron fluxes. These are clearly set by the available irradiation facilities.
The duration of the irradiation time. This is set by practical aspects, such as the limitations in total irradiation dose of the plastic containers because of radiation damage. The maximum irradiation time for polyethylene capsules is usually limited to several hours, for instance 5 hours at 5 × 1017 m-2s-1.
The total induced radioactivity that can be measured is set by the state-of-the-art of counting and signal processing equipment, with additional radiation dose and shielding considerations. As an example, the maximum activity at the moment of counting may have to be limited to approximately 250 kBq.
The duration of the counting time. A very long counting time may set limits to the number of samples processed simultaneously in case the radioactivity decays considerably during this counting time. Moreover, it reduces sample throughput.
The total turn-around time. Although sometimes better detection limits may be obtained at long decay times, the demands regarding the turn-around time often imply that a compromise has to be found between the longest permissible decay time and customer satisfaction.
The detector size, counting geometry and background shielding. The detector's characteristics may be set in advance by availability but several options exist.
It all illustrates that the detection limit for a given element by INAA may be different for each individual type of material, and analysis conditions. In Table 5 are given, as an indication, typical detection limits as derived from the analysis of a plant and a soil material. Peter Bode in his PhD thesis, Instrumental and organizational aspects of a neutron activation analysis laboratory, the typical detection limits as derived from the analysis of a plant and a soil material given in table 5 [26].
| NA | 2 | 10 | Nd | 0.7 | 8 | Ag | 0.2 | 2 |
| Ca | 700 | 4000 | Eu | 0.006 | 0.05 | Sn | 10 | 20 |
| Cr | 1 | 1 | Yb | 0.03 | 0.2 | Te | 0.3 | 3 |
| Co | 0.02 | 0.3 | Hf | 0.01 | 0.1 | Ba | 10 | 40 |
| Zn | 0.4 | 6 | W | 0.3 | 1 | Ce | 0.2 | 1 |
| As | 0.2 | 0.8 | Os | 0.1 | 0.6 | Sm | 0.01 | 0.03 |
| Br | 0.3 | 0.8 | Au | 0.003 | 0.01 | Tb | 0.008 | 0.1 |
| Sr | 5 | 60 | Th | 0.01 | 0.1 | Lu | 0.004 | 0.02 |
| Mo | 4 | 10 | K | 200 | 1500 | Ta | 0.01 | 0.2 |
| Cd | 3 | 8 | Sc | 0.001 | 0.02 | Re | 0.08 | 0.2 |
| Sb | 0.02 | 0.2 | Fe | 8 | 100 | Ir | 0.0006 | 0.004 |
| Cs | 0.02 | 0.3 | Ni | 2 | 30 | Hg | 0.05 | 0.4 |
| La | 0.1 | 0.3 | Ga | 2 | 10 | U | 0.2 | 2 |
| Se | 0.1 | 1 | Rb | 0.4 | 6 | Zr | 5 | 80 |
Table 5.
Detection limits of elements in mg.kg-1 as observed in NAA procedure of plant material and a soil material.
3. Applications
It is hardly possible to provide a complete survey of current NAA applications; however, some trends can be identified [27]. At specialized institutions, NAA is widely used for analysis of samples within environmental specimen banking programmes [28]. The extensive use of NAA in environmental control and monitoring can be demonstrated by the large number of papers presented at two symposia organized by the IAEA in these fields: "Applications of Isotopes and Radiation in Conservation of the Environment" in 1992 [29] and "Harmonization of Health-Related Environmental Measurements Using Nuclear and Isotopic Techniques" in 1996 [30]. Similar trends can also be identified from the topics discussed at the regular conference on “Modern Trends in Activation Analysis (MTAA)” and at the symposia on "Nuclear Analytical Methods in the Life Sciences" [31-33]. Additional sources of recent information on utilizing NAA in selected fields, such as air pollution and environmental analysis, food, forensic science, geological and inorganic materials as well as water analysis can be found in the bi-annual reviews in Analytical Chemistry, for instance cf. Refs [34-42]. It follows from these reviews that NAA has been applied for determining many elements, usually trace elements, in the following fields and sample types:
Archaeology: samples and objects such as amber, bone, ceramics, coins, glasses, jewellery, metal artefacts and sculptures, mortars, paintings, pigments, pottery, raw materials, soils and clays, stone artefacts and sculptures can be easily analyzed by NAA.
Biomedicine: the samples and objects that can be analysed include: animal and human tissues activable tracers, bile, blood and blood components, bone, brain cell components and other tissues, breast tissue, cancerous tissues, colon, dialysis fluids, drugs and medicines, eye, faeces, foetus, gallstones, hair, implant corrosion, kidney and kidney stones, liver, lung, medical plants and herbs, milk, mineral availability, muscle, nails, placenta, snake venom, rat tissues (normal and diseased), teeth, dental enamel and dental fillings, thyroid, urine and urinary stones.
In this work, we have used the INAA technique to analyse the traditional medicinal seeds prescribed for specific treatment purposes, were purchased from local markets [43]. The samples were irradiated at Es-Salam research reactor, at a power of 5 MW for 6 h. The accuracy of the method was established by analyzing reference materials. Twenty elements were measured, with good accuracy and reproducibility (Table 6). The concentration of elements determined, was found to vary depending on the seeds (Fig.11). The daily intake of essential and toxic elements was determined, and compared with the recommended values. The probable cumulative intake of toxic elements is well below the tolerance limits.
| Ba | mg/Kg | 7.7 ± 5.5 | 100.3 ± 5.8 | 112.4 ± 6.5 |
| Br | mg/Kg | 136.9 ± 4.6 | 119.6 ± 3.9 | 72.9 ± 2.4 |
| Ca | g/Kg | 3.77 ± 4.55 | 3.14 ± 0.39 | 1.50 ± 0.21 |
| Ce | mg/Kg | 1.44± 0.07 | 2.6 ± 0.1 | 1.98 ± 0.11 |
| Co | mg/Kg | 0.66 ± 0.02 | 0.73 ± 0.02 | 0.81 ± 0.03 |
| Cr | mg/Kg | 4.44 ± 0.19 | 29.3 ± 1.0 | 2.96 ± 0.20 |
| Cs | mg/Kg | 0.25 ± 0.01 | 0.51 ± 0.02 | 0.22 ± 0.01 |
| Eu | mg/Kg | 0.022 ± 0.002 | 0.039 ± 0.002 | 0.023 ± 0.002 |
| Fe | mg/Kg | 656.2 ± 71.6 | 823.2 ± 89.8 | 674.67 ± 74.16 |
| K | g/Kg | 3.67± 1.79 | 3.75 ± 0.20 | 3.7 ± 0.2 |
| La | mg/Kg | 0.74 ± 0.04 | 1.53 ± 0.06 | 1.50 ± 0.06 |
| Na | mg/Kg | 1028 ± 34 | 804.20 ± 26.69 | 615.50 ± 20.41 |
| Rb | mg/Kg | 24 ± 2 | 36.8 ± 1.4 | 26.3 ± 1.9 |
| Sc | mg/Kg | 0,258 ± 0,037 | 0,362 ± 0,051 | 0,272 ± 0,008 |
| Se | mg/Kg | 0,29 ± 0,04 | ND | ND |
| Sm | mg/Kg | 0,092 ± 0,004 | 0,18 ± 0,01 | 0,142 ± 0,005 |
| Sr | mg/Kg | 203,2 ± 7,8 | 136,88 ± 7,4 | 101,7 ± 4,7 |
| Th | mg/Kg | 0,159 ± 0,009 | 0,32 ± 0,02 | 0,195 ± 0,014 |
| Zn | mg/Kg | 68,06 ± 2,11 | 42,8 ± 1,4 | 40,24 ± 1,30 |
Table 6.
Elemental concentrations in the medicinal seed samples (Black seeds, Fenugreek, Caraway).
Environmental: in this domain, related fields concerned by NAA are: aerosols, atmospheric particulates (size fractionated), dust, fossil fuels and their ashes, flue gas, animals, birds, insects, fish, aquatic and marine biota, seaweed, algae, lichens, mosses, plants, trees (leaves, needles, tree bark), household and municipal waste, rain and horizontal precipitations (fog, icing, hoarfrost), soils, sediments and their leachates, sewage sludges, tobacco and tobacco smoke, surface and ground waters, volcanic gases.
Recently, our laboratory is strongly involved in various areas of application of k0-NAA. The present work focuses on the application of the k0-NAA method in Nutritional and Health-Related Environmental field [44]. Tobacco holds a leading position among different commodities of human consumption. The adverse health effects of toxic and trace elements in tobacco smoke on smokers and non-smokers are a special concern. In the present study, the concentration of 24 trace elements in cigarette tobacco of five different brands of Algerian and American cigarettes have been determined by k0-based INAA method. The results were compared with those obtained for samples from Iranian, Turkish, Brazilian and Mexican cigarettes tobacco. To evaluate the accurate of the results the SRM IAEA-140/TM was executed.
A multi-element analysis procedure based on the k0-NAA method was developed at Es-Salam research reactor allowing to simultaneously determine concentrations for 24 elements (As, Ba, Br, Ca, Ce, Co, Cr, Cs, Eu, Fe, Hf, K, La, Na, Rb, Sb, Sc, Se, Sm, Sr, Ta, Tb, Th, Zn). The determination of toxic and trace elements in cigarette tobacco is important both from the point of view of health studies connected with smoking and more general aspects of the uptake of trace elements by plants (table 7). Because of its great sensitivity, k0-NAA method is very suitable for determination of heavy metals such as As, Sb, Se and Zn. The accuracy of the results was checked by the analysis of standard reference material and good agreement was obtained with certified or literature values. The results of Algerian tobacco (table 8) were compared with analyses of Turkey [45], Iran [46], Mexican [47] and Brazilian tobacco [48].
| As | 4.05 ± 0.16 | 6.4 ± 0.24 | 2.16 ± 0.09 | 2.42 ± 0,09 | |
NSF Product and Service Listings
More than meets the eye: use of computer vision algorithms to identify stone tool material through the analysis of cut mark micro-morphology
References
Abellán N, Jiménez-García B, Aznarte J et al (2021) Deep learning classification of tooth scores made by different carnivores: achieving high accuracy when comparing African carnivore taxa and testing the hominin shift in the balance of power. Archaeol Anthropol Sci 13. https://doi.org/10.1007/s12520-021-01273-9
Adrian R (2017) Deep learning for computer vision with python - starter bundle. PyImageSearch. https://www.pyimagesearch.com/deep-learning-computer-vision-python-book/. Accessed 16 Apr 2021
Attallah O (2021) MB-AI-His: histopathological diagnosis of pediatric medulloblastoma and its subtypes via AI. Diagnostics 11:359. https://doi.org/10.3390/diagnostics11020359
Article Google Scholar
Ballard W (2018) Hands-on deep learning for images with TensorFlow: build intelligent computer vision applications using TensorFlow and Keras. Packt, Mumbai
Behrensmeyer AK, Gordon KD, Yanagi GT (1986) Trampling as a cause of bone surface damage and pseudo-cutmarks. Nature 319:768–771
Article Google Scholar
Bello SM (2010) New results from the examination of cut-marks using three-dimensional imaging. In: Ashton NM, Lewis SG, Stringer CB (eds) The ancient human occupation of Britain. Elsevier B.V, London, pp 249–262
Google Scholar
Bello SM, Soligo C (2008) A new method for the quantitative analysis of cutmark micromorphology. J Archaeol Sci 35:1542–1552. https://doi.org/10.1016/j.jas.2007.10.018
Article Google Scholar
Bello SM, Parfitt SA, Stringer C (2009) Quantitative micromorphological analyses of cut marks produced by ancient and modern handaxes. J Archaeol Sci 36:1869–1880. https://doi.org/10.1016/j.jas.2009.04.014
Article Google Scholar
Bonney H (2014) An investigation of the use of discriminant analysis for the classification of blade edge type from cut marks made by metal and bamboo blades. Am J Phys Anthropol 154:575–584. https://doi.org/10.1002/ajpa.22558
Article Google Scholar
Braun DR, Pobiner BL, Thompson JC (2008) An experimental investigation of cut mark production and stone tool attrition. J Archaeol Sci 35:1216–1223. https://doi.org/10.1016/j.jas.2007.08.015
Article Google Scholar
Braun DR, Pante M, Archer W (2016) Cut marks on bone surfaces: influences on variation in the form of traces of ancient behaviour. Interface Focus 6.https://doi.org/10.1098/rsfs.2016.0006
Chetlur S, Woolley C, Vandermersch P et al (2014) cuDNN: efficient primitives for deep learning. arXiv 1–9
Choi K, Driwantoro D (2007) Shell tool use by early members of Homo erectus in Sangiran, central Java, Indonesia: cut mark evidence. J Archaeol Sci 34:48–58. https://doi.org/10.1016/j.jas.2006.03.013
Article Google Scholar
Chollet F (2017) Deep learning with Python. Manning Publications Co., Shelter Island
Google Scholar
Cifuentes-Alcobendas G, Domínguez-Rodrigo M (2019) Deep learning and taphonomy: high accuracy in the classification of cut marks made on fleshed and defleshed bones using convolutional neural networks. Sci Rep 9:1–12. https://doi.org/10.1038/s41598-019-55439-6
Article Google Scholar
Courtenay LA, Yravedra J, Mate-González MÁ et al (2017) 3D analysis of cut marks using a new geometric morphometric methodological approach. Archaeol Anthropol Sci 11:651–665. https://doi.org/10.1007/s12520-017-0554-x
Article Google Scholar
Domínguez-Rodrigo M (2012) Stone tools and fossil bones. Cambridge University Press, Cambridge
Book Google Scholar
Domínguez-Rodrigo M, Cifuentes-Alcobendas G, Jiménez-García B et al (2020) Artificial intelligence provides greater accuracy in the classification of modern and ancient bone surface modifications. Sci Rep 10:1–12. https://doi.org/10.1038/s41598-020-75994-7
Article Google Scholar
Domínguez-Rodrigo M, Fernández-Jaúregui A, Cifuentes-Alcobendas G, Baquedano E (2021) Use of generative adversarial networks (Gan) for taphonomic image augmentation and model protocol for the deep learning analysis of bone surface modifications. Appl Sci 11(11). https://doi.org/10.3390/app11115237
Galán AB, Domínguez-Rodrigo M (2014) Testing the efficiency of simple flakes, retouched flakes and small handaxes during butchery. Archaeometry 56:1054–1074. https://doi.org/10.1111/arcm.12064
Article Google Scholar
Gifford-Gonzalez D (1991) Bones are not enough: analogues, knowledge, and interpretive strategies in zooarchaeology. J Anthropol Archaeol 10:215–254. https://doi.org/10.1016/0278-4165(91)90014-O
Article Google Scholar
Goodfellow I, Bengio Y, Courville A (2016) Deep learning. MIT Press, Massachussets
Google Scholar
Greenfield HJ (1999) The origins of metallurgy: distinguishing stone from metal cut-marks on bones from archaeological sites. J Archaeol Sci 26:797–808
Article Google Scholar
Greenfield HJ (2006) Slicing cut marks on animal bones: diagnostics for identifying stone tool type and raw material. J F Archaeol 31:147–163
Article Google Scholar
Jiménez-García B, Aznarte J, Abellán N et al (2020) Deep learning improves taphonomic resolution: high accuracy in differentiating tooth marks made by lions and jaguars. J R Soc Interface 17.https://doi.org/10.1098/rsif.2020.0446rsif20200446
Kingma DP, Ba JL (2015) Adam: a method for stochastic optimization. 3rd Int Conf Learn Represent ICLR 2015 - Conf Track Proc 1–15
Maté González MÁ, Yravedra J, González-Aguilera D, Palomeque-González JF, Domínguez-Rodrigo M (2015) Micro-photogrammetric characterization of cut marks on bones. J Archaeol Sci 62:128–142. https://doi.org/10.1016/j.jas.2015.08.006
Article Google Scholar
Maté-González MÁ, Palomeque-González JF, Yravedra J, González-Aguilera D, Domínguez-Rodrigo M (2016) Micro-photogrammetric and morphometric differentiation of cut marks on bones using metal knives, quartzite, and flint flakes. Archaeol Anthropol Sci 10:805–816. https://doi.org/10.1007/s12520-016-0401-5
Article Google Scholar
Merritt SR (2012) Factors affecting Early Stone Age cut mark cross-sectional size: Implications from actualistic butchery trials. J Archaeol Sci 39:2984–2994. https://doi.org/10.1016/j.jas.2012.04.036
Article Google Scholar
Misra D (2019) Mish: a self regularized non-monotonic neural activation function. arXiv
Olsen SL (1988) The identification of stone and metal toolmarks on bone artifacts. In: Olsen SL (ed) Scanning electron microscopy in archaeology. BAR International Series, London, pp 337–360
Chapter Google Scholar
Pizarro-Monzo M, Domínguez-Rodrigo M (2020) Dynamic modification of cut marks by trampling: temporal assessment through the use of mixed-effect regressions and deep learning methods. Archaeol Anthropol Sci 12.https://doi.org/10.1007/s12520-019-00966-6
Ramachandran P, Zoph B, Le QV (2017a) Searching for activation functions. arXiv 1–13
Ramachandran P, Zoph B, Le QV (2017b) SWISH: a self-gated activation function. arXiv 1–12
Rokach L (2010) Ensemble-based classifiers. Artif Intell Rev 33:1–39. https://doi.org/10.1007/s10462-009-9124-7
Article Google Scholar
Schmidhuber J (2015) Deep Learning in neural networks: an overview. Neural Netw 61:85–117. https://doi.org/10.1016/j.neunet.2014.09.003
Article Google Scholar
Val A, Costamagno S, Discamps E et al (2017) Testing the influence of stone tool type on microscopic morphology of cut-marks: experimental approach and application to the archaeological record with a case study from the Middle Palaeolithic site of Noisetier Cave (Fréchet-Aure, Hautes-Pyrénées, Franc. J Archaeol Sci Rep 11:17–28. https://doi.org/10.1016/j.jasrep.2016.11.028
Article Google Scholar
Von Lettow-Vorbeck CL (1998) El Soto de Medinilla: Faunas de mamíferos de la Edad del Hierro enel Valle del Duero (Valladolid, España). Archaeofauna 7:11–210
Google Scholar
Walker PL, Long JC (1977) An experimental study of the morphological characteristics of tool marks. Am Antiq 42:605–616
Article Google Scholar
West JA, Louys J (2007) Differentiating bamboo from stone tool cut marks in the zooarchaeological record, with a discussion on the use of bamboo knives. J Archaeol Sci 34:512–518. https://doi.org/10.1016/j.jas.2006.06.007
Article Google Scholar
Wolpert DH (1996) The lack of a priori distinctions between learning algorithms. Neural Comput 8:1341–1390. https://doi.org/10.1162/neco.1996.8.7.1341
Article Google Scholar
Wolpert DH, Macready WG (1997) No free lunch theorems for optimization. Trans Evol Comput 1:67–82. https://doi.org/10.1007/978-3-662-62007-6_12
Article Google Scholar
Yravedra J, Maté-González MÁ, Palomeque-González JF et al (2017) A new approach to raw material use in the exploitation of animal carcasses at BK (Upper Bed II, Olduvai Gorge, Tanzania): a micro-photogrammetric and geometric morphometric analysis of fossil cut marks. Boreas 46:860–873. https://doi.org/10.1111/bor.12224
Article Google Scholar
Download references
Acknowledgements
We want to thank the two reviewers of this manuscript for their useful insights in improving it to its final form. GCA also wants to thank Aimée Little for the help provided reviewing and imporving an earlier version of this paper.
Funding
Open Access funding provided thanks to the CRUE-CSIC agreement with Springer Nature. This study was funded by the Spanish Ministry of Economy and Competitiveness through the project (HAR2017-82463-C4-1-P) and the Ministry of Culture through their Archaeology Abroad program. Financial support has also been obtained from the Palarq Foundation and ESIN2.
Author information
Authors and Affiliations
Institute of Evolution in Africa (IDEA), Alcalá University, Covarrubias 36, 28010, Madrid, Spain
Gabriel Cifuentes-Alcobendas & Manuel Domínguez-Rodrigo
Area of Prehistory (Department History and Philosophy), University of Alcalá, 28801, Alcalá de Henares, Spain
Gabriel Cifuentes-Alcobendas & Manuel Domínguez-Rodrigo
Contributions
GCA created the experimental sample; GCA and MDR developed the python environment for CNNs; GCA and MDR carried out the analysis; GCA and MDR wrote and reviewed the manuscript.
Corresponding author
Correspondence to Gabriel Cifuentes-Alcobendas.
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
Reprints and Permissions
About this article

Cite this article
Cifuentes-Alcobendas, G., Domínguez-Rodrigo, M. More than meets the eye: use of computer vision algorithms to identify stone tool material through the analysis of cut mark micro-morphology. Archaeol Anthropol Sci13, 167 (2021). https://doi.org/10.1007/s12520-021-01424-y
Download citation
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s12520-021-01424-y
Share this article
Anyone you share the following link with will be able to read this content:
Sorry, a shareable link is not currently available for this article.
Provided by the Springer Nature SharedIt content-sharing initiative
Keywords
- Artificial intelligence
- Computer vision
- Raw material
- Cut marks
- Stone tools
- Palaeolithic
Role of Natural Stone Wastes and Minerals in the Alkali Activation Process: A Review
Bartolomeo Coppola,1Jean-Marc Tulliani,1Paola Antonaci,2,3,* and Paola Palmero1
Paola Antonaci
2Department of Structural, Geotechnical and Building Engineering, Politecnico di Torino, Corso Duca Degli Abruzzi, 24, 10129 Torino, Italy
3Responsible Risk Resilience Centre, Politecnico di Torino, Viale Mattioli 39, 10125 Torino, Italy
Find articles by Paola Antonaci
Author informationArticle notesCopyright and License informationDisclaimer
1INSTM R.U. Lince Laboratory, Department of Applied Science and Technology, Politecnico di Torino, Corso Duca Degli Abruzzi, 24, 10129 Torino, Italy; ti.otilop@aloppoc.oemolotrab (B.C.); ti.otilop@inaillut.cramnaej (J.-M.T.); ti.otilop@oremlap.aloap (P.P.)
2Department of Structural, Geotechnical and Building Engineering, Politecnico di Torino, Corso Duca Degli Abruzzi, 24, 10129 Torino, Italy
3Responsible Risk Resilience Centre, Politecnico di Torino, Viale Mattioli 39, 10125 Torino, Italy
*Correspondence: ti.otilop@icanotna.aloap
Received 2020 Mar 24; Accepted 2020 May 9.
Abstract
This review aims to provide a comprehensive assessment concerning alkali activation of natural stone wastes and minerals. In particular, the structure of the review is divided into two main sections in which the works dealing with alumino-silicate and carbonatic stones are discussed, respectively. Alumino-silicate stones are generally composed of quartz and feldspars, while carbonatic stones are mainly made of calcite and dolomite. The role of these minerals in the alkali activation process is discussed, attesting their influence in the development of the final product properties. In most of the works, authors use mineral additions only as fillers or aggregates and, in some cases, as a partial substitution of more traditional raw powders, such as metakaolin, fly ash, and granulated blast furnace slag. However, a few works in which alumino-silicate and carbonatic stone wastes are used as the main active components are discussed as well. Not only the raw materials, but also the entire alkali activation process and the curing conditions adopted in the literature studies here reviewed are systematically analyzed to improve the understanding of their effect on the physical, mechanical, and durability properties of the final products and to eventually foster the reuse of natural stone wastes for the purposes of sustainability in different applications.
Keywords: alkali activation, geopolymer, stone waste, stone, minerals, alumino-silicate, carbonates, calcite, dolomite, reuse, sustainability
1. Introduction
The interest towards geopolymers and alkali-activated materials arises due to the search for lower carbon dioxide (CO2) binders as compared to traditional cement [1,2,3,4,5].
The term ‘alkali-activated materials’ normally refers to aluminate-rich materials showing cementitious properties as a consequence of reactions initiated by an alkaline activator. However, as underlined by J. Davidovits [6], geopolymers and alkali-activated materials are characterized by significantly different structures, compositions, and properties. The former, in fact, are generated by polymer chemistry mechanisms (poly-sialate); the latter are just hydrated and/or precipitated products [7]. More in detail, geopolymer formation occurs via dissolution of silica (SiO4) and alumina (AlO4) tetrahedra and subsequent condensation phenomena, giving rise to larger alumino-silicate oligomers, which further condense to form large structural units [8]. In such a way, a three-dimensional polymer structure is formed, and this arrangement accounts for the high durability of geopolymers, which is generally superior to that of alkali-activated materials [6,7].
Traditionally, geopolymers are produced starting from metakaolin (obtained by the high-temperature calcination of kaolin [9]) as a source of Al and Si species, as well as an alkaline activator. The latter is a mixture of sodium or potassium hydroxide (NaOH or KOH) aqueous solution, added with liquid sodium silicate. Therefore, the geopolymer development requires the consumption of natural resources (both clay and the raw materials necessary to produce the chemical activators) and their high-energy treatment, so this process is associated with great environmental impacts [10,11,12].
The keys to reducing the carbon footprint associated with this process rely on the search for alternative raw materials and on the reduction of the amount and concentration of the alkaline activator, or at least of one of its components [11]. The most investigated strategy implies the use of specific industrial waste as an alternative alumina-silicate source, such as fly ash, ground granulated blast furnace slag, and various types of slags [10,12,13,14]. As a common feature, these industrial wastes are characterized not only by high silicate and aluminate contents, but also by their predominantly amorphous or poorly crystallized structure. This makes these materials highly reactive when exposed to the alkaline activator, thus favoring the dissolution and condensation reactions necessary to generate high-strength hardened materials.
A more innovative approach implies the exploitation of industrial mineral wastes with highly crystalline structures, such as those produced during the extraction and processing of ornamental stones. In fact, huge volumes of natural stones are extracted worldwide, corresponding to impressive amounts of related wastes [15]. As an example, Figure 1 reports some pictures of typical installations of alumino-silicate- and carbonate-based ornamental stones, as well as pictures of the production process and resulting waste powders. With reference to marble and granite production, the world dimension is estimated to be approximatively 155 million tons (2014 data); China, India, Turkey, Iran, and Italy are the top five productive countries, which account for about 74% of the total world stone production dimension [16]. Moreover, this market, especially in Europe, is continuously increasing at an annual rate of 7% [17]. The stone manufacturing chain implies three main activities, here specified: (i) Extraction from quarry, (ii) finishing treatments (such as cutting, smoothing, and polishing), and (iii) transportation and sale [18]. These activities are associated with important landscape and environmental impacts, particularly during the extraction and cutting phases. During extraction, around 30% of the stone goes to scrap because of small size and/or irregular shape [19,20]. Moreover, large quantities of stone fines (named quarry dust) are generated, which could be harmful if dispersed in air, water, or soil [15]. On the other hand, the cutting and polishing phases produce a large amount of sludge, which is essentially constituted by a mix of quarry dusts and cooling water used in the working process [18]. It can be estimated that one ton of marble stone processed in a gang-saw or vertical/horizontal cutter produces almost one ton of slurry, 70% of which is water [20]. The water content is normally reduced by press filtration, thus producing a mud with a water content lower than 20% [21]. Generally, this waste is landfilled, while, in some cases, it is dumped directly into the ecosystem. Such improper disposal can be hazardous for the environment, producing soil and water contamination as well as necrotic conditions for flora and fauna. One concern is related to the fine particle size and the lack of pores, making the mud almost impermeable to oxygen and causing asphyxia in living organisms [22]. If a large amount of dust is dispersed in water, it can increase the water’s turbidity. As a consequence, light penetration is inhibited and photosynthesis activity is reduced, causing a lack of nutrients and modification of the food chain [23,24]. Finally, the dried mud and fine dust can be easily dragged by wind, becoming harmful to humans and animals through inhalation, inducing asthma, silicosis, and lung cancer [24]. A last issue concerns the possible presence of heavy metals in sludge due to cutting operations using frame saws. However, this concern is nowadays almost overcome thanks to the increasing use of metal-free diamond cutting discs.
In addition to the previously described health and environmental issues, quarry waste landfilling is very costly, corresponding to more than 3% of the operating costs of stone working plants [22]. Therefore, the incorporation of these wastes in other industrial processes could generate cost reduction and new business opportunities, while reducing the extraction of raw materials and preserving natural resources.
In light of this, the use of ornamental stone waste in the construction industry could be a smart solution to avoid landfilling and dust propagation in the environment. A recent work by Galetakis and Soultana [15] reviewed the use of quarry fines in the preparation of building materials. Authors found that slurries can be used as fine aggregates or as cement replacement [25,26,27], while solid waste can, in part, substitute aggregates in concrete mixtures, as already proposed in literature for other wastes as well [28,29,30,31,32].
More recently, the possibility of including fine quarry wastes into new alkali-activated products has been investigated, similarly to what other researchers are doing as regards alkali-activated materials containing mine tailings (i.e., the finely ground residue from ore extraction) [33,34,35,36]. The aim of this review, in fact, is to highlight the recent advances achieved by applying the alkaline activation approach to the finest fraction of the quarry wastes that have either a siliceous or carbonate nature. A summary of the papers discussed in this review is presented in Table 1. First, a classification based on the chemical nature of the waste is done. In the alumino-silicate class, a variety of mineral wastes are used, such as granite, albite, pietra serena, pumice, andesite, etc. The choice of alumino-silicate waste for the alkali activation process is based on the chemical similarity of these minerals to the commonly used raw materials, in which precursors rich in Si+Al and low in Ca contents give rise to inorganic polymers of sodium aluminum silicate hydrate (N-A-S-H) of high mechanical strength [37,38]. Concerning carbonates, the main exploited wastes derive from calcite, dolomite, marble, and limestone processing. Here, a possible role played by calcium on alkali-activated products is taken into consideration, since the formation of a calcium silicate hydrate (C-S-H) gel has been postulated in some previous works, with significant effects on the materials’ mechanical properties and durability [39,40,41,42,43,44]. It is interesting to observe from Table 1 that most publications deal with the use of mineral muds/dusts as additives to common geopolymer sources, such as metakaolin, fly ash, and granulated blast furnace slag. In fact, due to the highly crystalline nature of stone waste, its reactivity under alkaline conditions is significantly reduced, thus requiring an ‘active’ material to provide the setting and hardening stages and to control the whole process. However, very few studies rely on the exploitation of such very fine wastes as primary sources for alkali-activated materials [45,46]. To complete the overview of the raw materials, the chemical compositions of the alumino-silicate and carbonate materials used in the reviewed papers are reported in detail in Table 2 and Table 3, respectively.
Table 1
Summary of the papers related to the use of stone waste/minerals in alkali activation processes. A classification of the minerals based on their chemical nature (alumino-silicate or carbonate) is provided, in addition to the composition of the ‘active’ raw materials for alkali activation (if any) and the general composition of the liquid alkaline activator.
| Classification | Mineral Additive | Alkali-Activation Source | Activator | Reference |
|---|---|---|---|---|
| Alumino-silicates | Granite | Fly Ash/Granulated Blast Furnace Slag | NaOH + Na2SiO3 + H2O | [47] |
| Granite | Metakaolin | NaOH + Na2SiO3 + H2O | [48] | |
| Granite | - | NaOH + Na2SiO3 + H2O | [46] | |
| Albite + | - | Na2CO3/NaOH | [49] | |
| Pietra Serena | Metakaolin | NaOH + Na2SiO3 + H2O | [50] | |
| Pisha sandstone | -/Fly Ash | NaOH + Na2SiO3 + H2O | [51] | |
| Pisha sandstone | - | NaOH + NaCO3 + Na2SO4 + Na2SiO3 + H2O | [52] | |
| Cordierite | Metakaolin | NaOH + Na2SiO3 + H2O | [53] | |
| Diatomite | - | NaOH + H2O + Wood Biomass Ash ^ | [54] | |
| Carbonates | Calcite | Metakaolin | NaOH + Na2SiO3 + H2O | [55] |
| Calcite | Granulated Blast Furnace Slag | NaOH + H2O | [56] | |
| Calcite | Metakaolin | NaOH/KOH + Na2SiO3 + H2O | [57] | |
| Calcite | Fly Ash | NaOH + Na2SiO3 + H2O | [58] | |
| Dolomite | Fly Ash/Granulated Blast Furnace Slag | Na2CO3/NaOH | [59] | |
| Dolomite | Metakaolin | NaOH + Na2SiO3 + H2O | [55] | |
| Dolomite + | Bentonite + Na2CO3 | H2O | [60] | |
| Dolomite + | Granulated Blast Furnace Slag + Na2CO3 | H2O | [61] | |
| Dolomite | Fly ash/cement | NaOH + Na2SiO3 + H2O | [62] | |
| Marble ° | - | NaOH + H2O | [63] | |
| Marble | - | NaOH + Na2SiO3 + H2O | [45] | |
| Marble | Smectite clay | NaOH + Na2SiO3/Sodium citrate + H2O | [64] | |
| Marble | Cement/Fly ash/GBFS/Gypsum/Clay | NaOH + Na2SiO3 + H2O | [65] | |
| Marble | Fly ash | NaOH + Na2SiO3 + H2O | [66] | |
| Pietra di Angera | Metakaolin | NaOH + Na2SiO3 + H2O | [50] | |
| Travertine ° | - | NaOH + H2O | [63] | |
| Limestone | Metakaolin | NaOH + H2O | [67] | |
| Limestone | Fly Ash/Granulated Blast Furnace Slag | NaOH + Na2SiO3 + H2O | [68] | |
| Limestone | Granulated Blast Furnace Slag | Na2CO3 + H2O | [69,70] | |
| Limestone | - | NaOH + Na2SiO3 + H2O | [40] | |
| Limestone | Halloysite clay | NaOH + Na2SiO3+H2O | [71] | |
| Limestone * | Granulated Blast Furnace Slag | NaOH + Na2CO3 + H2O | [72] | |
| Mixed | Dolomite + Microcline + Albite + Quartz | - | NaOH + Na2SiO3 + H2O | [46] |
| Marl + | - | NaOH + Na2SiO3 + H2O | [73] | |
| Marl ++Limestone | - | Na2SiO3 + H2O | [74] | |
| Marl/Marl + | Granulated Blast Furnace Slag | Na2SiO3 + H2O | [75] | |
| Pietra serena sludge | Fly ash/metakaolin | NaOH + H2O | [76] | |
| Garnet tailings | Metakaolin | Na2SiO3 + NaOH + H2O | [77] |
Open in a separate window
Table 2
Chemical composition according to X-ray fluorescence spectroscopy analyses (XRF), alkali-activating solution details, curing conditions, and compressive strength of alkali-activated materials containing alumino-silicate minerals.
| Mineral Additive (Chemical Composition) | Alkali Activation and Curing Regimen | Max. Compressive Strength + | Reference |
|---|---|---|---|
| Granite (62.11% SiO2; 15.72% Al2O3; 4.98% K2O) | 10 M NaOH | 30.5 MPa (10% of granite in FA-based geopolymers); 72.6 MPa (10% of granite in GBFS-based geopolymers) | [47] |
| 80 °C for 24 h | |||
| Granite (68.10% SiO2; 15.80% Al2O3; 5.32% K2O) | Ms * = 1.64 (H2O/Na2O molar ratio of 13) | 35 MPa | [46] |
| 80 °C for 24 h | |||
| Pietra Serena (59% SiO2; 16% Al2O3; 6.3% MgO) | 10, 14, 16 and 20 H2O/Na2O molar ratio (mixing H2O + Na2SiO3 + NaOH) | 21 MPa (H2O/Na2O molar ratio = 20; metakaolin:pietra Serena = 1:1) | [50] |
| 20 °C and 90% RH | |||
| Granite (60.51% SiO2; 17.49% Al2O3; 8.71% Fe2O3) | Na2SiO3 was used to activate fused granite (with several Ms*) and MK (added to balance Na2/Al2O3 ratio) | 40.5 MPa (for mortars containing fused granite wastes with SiO2/Na2O = 0.47 and Al2O3/Na2O = 0.08) | [48] |
| 24 h at room temperature closed in plastic bags, then 25 °C and 90% RH | |||
| Pisha sandstone (65.64% SiO2; 14.35% Al2O3; 8.02% CaO) | 0.7, 1.2, and 1.6 wt.% of NaOH with respect to 100 g of Pisha sandstone | 6 MPa (ambient cured and 1.2 wt.% NaOH) | [51] |
| 80 °C for 24 h then ambient and water immersed | |||
| Pisha sandstone (62.46% SiO2; 20.08% Al2O3; 5.10% CaO) | Na2SiO3 (Ms* = 1.5, 2.0 and 3.0); Na2CO3, Na2SO4; NaOH | 14.4 MPa (Na2SiO3 with Ms = 3, cured at 80 °C and milled Pisha stone) | [52] |
| (i) 80 °C for 24 h and (ii) ambient temperature | |||
| Cordierite (52.85% SiO2; 34.62% Al2O3; 11.66% MgO) | H2O/Na2O = 13, 15, and 20 | 57.5 MPa (30% of Cordierite; H2O/Na2O = 13) | [53] |
| sealed and room temperature | |||
| Diatomite (80.3% SiO2; 6.1% Al2O3; 6.79% Fe2O3) | 3 wt.% of NaOH; w/p = 0.27 | 48 MPa | [54] |
| 23 °C and 99% RH | |||
| Albite (70.9% SiO2; 17% Al2O3; 9.75% Na2O) | Albite calcined with NaOH or Na2CO3; w/s = 0.3 | 44.2 MPa (Albite calcined with 50% NaOH at 1000 °C) | [49] |
| sealed and room temperature |
Open in a separate window
Table 3
Chemical composition according to X-ray fluorescence spectroscopy analyses (XRF), alkali-activating solution details, curing conditions, and compressive strength of alkali-activated materials containing carbonate minerals.
| Mineral Additive (Chemical Composition) | Alkali-Activation and Curing Regimen | Max. Compressive Strength + | Reference |
|---|---|---|---|
| Calcite (53.5% CaO; 1.7% MgO; 1.5% SiO2) | Ms* = 2.0, 1.5, and 1.2 | ~60 MPa (Ms = 1.5, MK with 20% of calcite) | [55] |
| 40 °C for 24 h | |||
| Calcite (55.91% CaO; 0.18% K2O; 0.09% SiO2) | 2%, 4%, and 6% of NaOH | ~70 MPa (4% of NaOH, 5% of calcite, and 91% of GBFS) | [56] |
| 37 °C 100% RH | |||
| Calcite (purity 98.5%) | 13% KOH, 10% NaOH, 27% Na2SiO3, 50% H2O | ~28 MPa (MK with 6% of calcite) | [57] |
| 40 °C for 12 h | |||
| Calcite (50.1% CaO; 3.9% SiO2; 1.7% Al2O3) | 3, 6, and 12 M NaOH | 19.2 MPa (12 M, 67% mineral addition, 5 h curing) | [58] |
| 80 °C for 1, 3, and 5 h | |||
| Marble (53.68% CaO; 1.32% Fe2O3; 0.26% SiO2) | 1, 5, and 10 M NaOH | 37.48 MPa (10 M NaOH, curing 20 °C, 20% Marble) | [63] |
| (1) 22 °C and 40% RH; (2) 45 °C for 24 h; (3) 75 °C for 24 h. Afterwards, wet condition (min. 95% RH) or 22 °C and 35% RH | |||
| Marble (55.9% CaO; 0.6% MgO; 0.1% Fe2O3) | (1) 0.1 mol of Na2O; (2) 0.1 mol of Na2O and 0.1 mol SiO2; (3) substitution of sodium citrate (Na3C6H5O7) | 60.7 MPa (25% Marble/75% calcined smectite clay; containing Na-citrate) | [64] |
| 24 h at 95% RH then dry-cured at room temperature | |||
| Marble (44.20% CaO; 1.41% MgO; 0.07% Fe2O3) | Ms* = 1.65 and 3.50 | 38.30 MPa (Ms = 1.65, dry curing) | [45] |
| 24 h at 80 °C then: (1) 20 °C and 95% RH; (2) 20 °C and 18% RH; (3) immersed in water | |||
| Marble (45.60% CaO; 6.82% MgO; 0.70% SiO2) | 8 M NaOH or Na2SiO3·nH2O:NaOH (w:w = 5) | 52 MPa (Na2SiO3·nH2O + NaOH; cement, GBFS, marble, FA) | [65] |
| Room temperature | |||
| Marble (38.02% CaO; 34.66% SiO2; 13.12% Fe2O3; 7.21% MgO) | 2 and 4 M NaOH + Na2SiO3 | 6.52 MPa (7 days, 4 M, 60% marble + 40% FA) | [66] |
| 70 °C for 24 h and 7 days in plastic bags | |||
| Travertine (55.10% CaO; 0.70% SiO2; 0.20% Fe2O3) | 1, 5, and 10 M NaOH | 42.24 MPa (10 M NaOH, dry curing at 20 °C, 20% travertine) | [63] |
| (1) 22 °C and 40% RH; (2) 45 °C for 24 h; (3) 75 °C for 24 h. Afterwards, wet condition (min. 95% RH) or 22 °C and 35% RH | |||
| Dolomite (33.4% CaO; 17.1% MgO; 2.5% SiO2) | Ms* = 2.0, 1.5, and 1.2 | ~45 MPa (Ms = 1.5, MK with 20% of dolomite) | [55] |
| 40 °C for 24 h | |||
| Dolomite (42.48% CaO, 19.15% MgO) | Ms* = 2.5 (Na2CO3 and bentonite) | 38.3 MPa (bentonite, dolomite and Na2CO3 calcined at 1110 °C and (CaO + MgO)/SiO2 = 2.1) | [60] |
| 80 °C for 3 days | |||
| Dolomite (74.8% CaO; 18.3% MgO; 3.7% SiO2) | Ms*= 0.93 (10 M NaOH + Na2SiO3) | ~60 MPa (Cement with 40% of dolomite); ~40 MPa (FA with 40% of dolomite); | [62] |
| Cement-based samples 24 h at 100% RH then immersed in lime water; FA-based samples 24 h at 40 °C and 100 % RH then sealed and at room temperature | |||
| Dolomite (31.4% CaO, 21.3% MgO, 1.1% SiO2) | Na2CO3 + calcined dolomite (Na2O = 4.9%–7.6% in the dry mixture) | 41.6 MPa (GBFS with 10% of calcined dolomite and 10% of Na2CO3) | [61] |
| 20 °C and RH > 95% ± 2% | |||
| Dolomite (27.13% CaO; 24.53% MgO; 0.13% SiO2) | 4 M NaOH or 2 M Na2CO3 | ~60 MPa (NaOH, GBFS and 20% of dolomite); ~80 MPa (Na2CO3, GBFS and 20% of dolomite); | [59] |
| 20 °C 100% RH | |||
| Pietra di Angera (64% CaO; 33% MgO; 2.2% Fe2O3) | 10, 14, 16, and 20 H2O/Na2O molar ratio (obtained mixing H2O + Na2SiO3 + NaOH) | 18 MPa (H2O/Na2O molar ratio = 20; MK:pietra di Angera = 1:1) | [50] |
| 20 °C and 90% RH | |||
| Limestone (53.5% CaO; 1.7% MgO; 1.5% SiO2) | 3 and 5 M NaOH | 7 MPa (50% LM/50% MK; 5 M NaOH; 20 °C; dry curing) | [67] |
| 24 h at 20 or 80 °C then wet curing (water immersed) or dry curing (laboratory conditions) | |||
| Limestone (53.96% CaO; 1.01% MgO; 0.84% SiO2) | Ms * = 1.4 | 83.5 MPa (30% LM/10% FA/60% GBFS) | [68] |
| 20 °C and 95% RH | |||
| Limestone (53.96% CaO; 1.01% MgO; 0.84% SiO2) | 4 wt.% of Na2O (starting from Na2CO3) | ~55 MPa (GBFS with 10% of LM) | [69,70] |
| 20 °C and 95% RH | |||
| Limestone (57.43% CaO; 1.06% SiO2) | Ms * = 0 (only NaOH), 1, and 1.5 | ~15 MPa (Ms = 1, 10% Na2O) | [40] |
| 24 h at 60 °C then dry stored in plastic bags at 20 °C and 80%–90% RH | |||
| Limestone (47.85% CaO; 9.07% SiO2; 1.51% Al2O3) | 5, 10, and 8 M NaOH + Na2SiO3 | 47.77 MPa (8 M, 45% LM/55% Clay) | [71] |
| 24 °C in open air | |||
| Limestone 1 (90% calcite, 9% quartz) (43.31% CaO; 14.26% SiO2; 2.44% Al2O3) | NaOH + Na2CO3 + H2O | 39 MPa (30% LM_3/70% GBFS; Ssp 600 m2/kg) | [72] |
| Limestone 2 (33% calcite, 66% dolomite) (39.79% CaO; 1.26% SiO2; 12.94% MgO) | |||
| Limestone 3 (100% calcite) (55.06% CaO; 0.47% SiO2; 0.49% MgO) | Room temperature and 95%–100% RH |
Open in a separate window
Following the classification reported in Table 1, alumino-silicate and carbonate-containing alkali-activated materials are discussed in separate sections of this review (Section 2 and Section 3, respectively) and, for each of them, the roles of composition and processing (Section 2.1 and Section 3.1) as well as the properties of fresh and hardened materials (Section 2.2 and Section 3.2) are reviewed. Then, the specific role exerted by both mineral fines on the alkali activation process is considered (Section 2.2 and Section 3.2), followed by a final discussion (Section 4) aimed to provide a scientific contribution for maximizing the use of these wastes in industry and consequently reducing the impacts related to the manufacturing of ornamental stones.
2. Alkali-Activated Materials Based on Alumino-Silicate Minerals
2.1. Role of Raw Materials, Activators, and Curing Conditions
Different natural alumino-silicate minerals have been investigated in the scientific literature in combination with traditional alkali-activated materials. In particular, granite [46,47,48], albite [49], Pietra serena [50], Pisha sandstone [51,52], cordierite [53], and diatomite [54] have been used in combination with metakaolin (MK) [48,50,53] and fly ash (FA) [47,51]. As previously mentioned, only a few works investigated the use of alumino-silicate stones alone [46,49,51,52,54], i.e., without any other ‘active’ component. Alumino-silicate stones are mainly composed of quartz (SiO2) and feldspars, such as microcline (KAlSi3O8) and albite (NaAlSi3O8). In some cases, some phyllosilicates belonging to the mica group, like biotite and muscovite, can also be present. As regards the oxides, these materials are mainly composed of silica (with values ranging between 50% and 70%) and alumina (between 15% and 35%), as reported in Table 2. A certain difference is presented by diatomite, which is a naturally occurring siliceous sedimentary rock that is characterized by a very high silica amount (80.3%) and a lower alumina content (6.1%) [54].
Clausi et al. [50] investigated the possibility of using an ornamental stone, i.e., Pietra Serena (an Italian sandstone) in combination with MK for the production of mortars. MK/Pietra Serena alkali-activated mortars can be used in restoration of cultural heritage because they mimic the natural stone, thus allowing aesthetic compatibility [17]. Crushed Pietra Serena was used for aggregate preparation, keeping the clayey fraction as well, which provided the necessary color shades for aesthetic reasons.
Tchadjié et al. [48] used granite waste from Cameroon in the preparation of MK-based geopolymers. In particular, the authors fused granite powder (at 550 °C for 2 h) with Na2O at different weight percentages (from 10 to 60 wt.%, 10 wt.% of increments). Then, fused granite powder (d < 90 μm) was mixed with MK and activated with Na2SiO3 solution [48].
Hemra and Aungkavattana [53] investigated the influence of cordierite (Mg2Al4Si2O18) addition, from 0 to 50 wt.%, into MK-based geopolymers.
Hassan et al. [54] investigated the possibility of preparing geopolymers by mixing wood biomass ash (WBA) and diatomite. Even if diatomite is a sedimentary siliceous rock, it is mainly composed of alumino-silicates rich in amorphous iron.
Choi et al. [47] replaced fly ash (FA) and granulated blast furnace slag (GBFS) with a stone powder sludge derived from granite quarries. The authors investigated several replacement ratios (10, 20, and 30 wt.%, respectively) using the stone sludge as received, i.e., with a high water content (20.7%) [47].
An original contribution is provided by Palmero et al. [46], who exploited a granite quarry mud as a unique feedstock material for alkali-activated products, and this technology was the object of two recent patent applications [78,79]. Due to the almost fully crystalline nature of the raw powder, the developed material represents a clear innovation in the field of alkali-activation processes [46]. Similarly, Li et al. [51,52] investigated the alkali activation of Pisha sandstone (a Chinese sandstone) both alone [51,52] and mixed with FA [51]. Pisha sandstone is mainly composed of crystalline minerals, i.e., quartz and albite, with SiO2 and Al2O3, accounting for approximatively 80 wt.% of the whole composition (Table 2). Feng et al. [49] investigated the possibility of activating pure albite, but it was previously thermally treated with sodium hydroxide or sodium carbonate. In particular, albite was mixed with different amounts (10%, 30%, and 50% with respect to albite mass) of sodium hydroxide or sodium silicate and then heated at four different temperatures (850, 900, 1000, and 1150 °C, respectively) for 30 min [49].
Details related to curing conditions and alkaline solutions of the different reviewed papers are reported in Table 2. It can be observed that, in some researches, the materials were cured at 80 °C [46,47,51,52], even if in most of the works, curing at room temperature was preferred [48,49,50,52,53,54].
In addition, the heat-curing was always performed for a short time (a few hours or a few days), followed by a longer room temperature curing. For example, in the studies reported in [46,47], specimens were cured at 80 °C for 24 h and then kept at 23 °C until testing.
The curing atmosphere plays a role, too. For instance, Li et al. [51] investigated different curing conditions for geopolymers prepared using a Chinese sandstone (i.e., Pisha sandstone) by exposing the samples at 80 °C for 24 h, followed by room temperature curing performed under air or water. Authors reported lower mechanical properties in the case of water-cured samples, probably due to excessive water, which hindered the polycondensation reactions [51].
A further important parameter that affects the performance of the fresh and hardened materials is the activating solution. Li et al. investigated several formulations: In [51], the authors kept Na2SiO3 and water content constant while varying NaOH concentration, thus obtaining solutions at three different SiO2/Na2O molar ratios (Ms), equal to 1.5, 2.0, and 3.0. They showed that the strength decreased by increasing Ms. On the contrary, in [52], the same authors tested different activators (Na2SiO3, Na2CO3, Na2SO4, and NaOH, respectively) in order to investigate the role of pH on the development of alkali-activated materials. They showed that the higher the pH, the stronger the materials.
A recent and interesting approach deals with the design and development of one-part geopolymers, which are mixtures of solid alumino-silicate precursors and solid alkaline chemicals; the addition of ‘just water’ is responsible for the activation, similarly to cement technology [80,81,82]. The term is in opposition to the traditional two-part geopolymers, in which the solid precursors are mixed with a liquid alkaline solution: Issues related to the high viscosity and pH of the solution make the scalability of geopolymers difficult, restricting them to relatively small-scale applications and pre-cast components. Two previous researches investigated the possibility of preparing one-part geopolymers with natural minerals [49,54]. In particular, Feng et al. [49] prepared alkali-activated materials starting from a thermally treated albite and adding only water. The authors fixed the water/solid ratio at 0.30 and cured samples at room temperature in sealed containers [49]. On the other side, Hassan et al. [54] prepared one-part geopolymers mixing a dry activator with diatomite. In particular, the dry activator was prepared mixing a CaCO3-rich ash (WBA) with a dried solution of sodium hydroxide. Then, diatomite was mixed with different amounts of the dry activator and activated with water at a water/powder ratio of 0.27 and 3 wt.% of NaOH. Samples were finally cured at 23 °C and 99% relative humidity (RH).
2.2. Properties of Fresh and Hardened Materials
2.2.1. Fresh Mixtures Properties
Properties of fresh mixtures are very important in alkali-activated materials not only for casting, but also for mechanical property development; in fact, water greatly influences workability and alkali activation reactions. In spite of this, only a few works studied the fresh properties of alkali-activated alumino-silicate mineral fines [46,49,54].
Palmero et al. [46] determined the apparent viscosity of alkali-activated granite pastes using a viscometer (in the range of 10–40 s−1) and found that values between 35 and 40 Pa·s were suitable for casting and mechanical property development.
Hassan et al. [54] studied initial and final setting times of one-part geopolymers made with a dry activator (WBA and NaOH) and diatomite. The authors measured an increase in setting time (both initial and final) with increasing dry activator content due to the dilution effect of calcium carbonate (contained in WBA) on the amount of amorphous reactive silica, and therefore on the formation of the binding phases [54]. Feng et al. [49] reported a setting time in the order of few minutes for one-part alkali-activated thermally treated albite. Indeed, this mixture containing alkalis (sodium hydroxide or sodium carbonate) was highly reactive, and a significant amount of heat was released after mixing with water [49].
2.2.2. Mechanical Properties of Hardened Materials
The influence of alumino-silicate powder addition on the mechanical properties of the final alkali-activated material is reviewed in the following, and the highest compressive strengths achieved in the previous researches are summarized in Table 2.
Choi et al. [47] obtained two opposite trends in replacing FA and GBFS with granite sludge. In particular, for FA, at increasing sludge replacement, the compressive strength decreased, even at the lowest replacement (10 wt.% sludge). On the contrary, for GBFS, 10 wt.% replacement produced an increase of compressive strength (from 61.7 to 72.6 MPa), while higher substitutions provided lower values, but still higher than the reference neat GBFS material.
Clausi at al. [50] compared the mechanical properties of alkali-activated MK mortars prepared using a standard sand or Pietra Serena crushed sand as aggregates. The authors reported a significant decrease of both flexural and compressive strength compared to the reference mortar; however, the compressive strength achieved (21 MPa) was still suitable for masonry mortars. The strength decrease was attributed to the different particle shape (angular morphology) and to the higher presence of fines in Pietra Serena sand with respect to the standard sand.
Tchadjié et al. [48] investigated the mechanical properties of fused granite waste/MK mortars at different Na2O amounts in fused granite waste. In particular, the authors reported a progressive increase of compressive strength up to a Na2O content of 40 wt.% followed by a strength decrease. This Na2O wt.% was found to be the most favorable in terms of geopolymerization degree and strength development.
Hemra and Aungkavattana [53] obtained an increase of compressive strength at increasing cordierite addition in MK-based geopolymers. In particular, a 26% compressive strength increase (from 42.5 to 57.5 MPa) was measured by adding 30 wt.% of cordierite. The authors explained this result in terms of filler effect, because cordierite particles acted as aggregate and avoided crack propagation, improving the mechanical properties. Thus, no influence on the geopolymerization process was attributed to cordierite.
Li et al. [51,52] explored alkali activation of a Chinese sandstone (i.e., Pisha sandstone) both alone and mixed with FA, and investigated the effects of several parameters. First, for pure Pisha sandstone, the authors stated an important role of curing conditions; the 28 day compressive strength of samples cured under air (~6 MPa) is higher than that of water cured materials (~4 MPa) [51]. Second, the curing time showed a role too, since, in both cases, higher mechanical properties were achieved after 90 days of curing. Third, the higher the NaOH content in the alkaline solution, the higher the compressive strength. Finally, if the sandstone was mixed with FA, the compressive strength further increased. In a following study, the same authors investigated the influence of the activator type and Pisha sandstone particle size [52]. Milled Pisha sandstone (mean diameter of 18.9 μm) yielded higher compressive strengths compared to un-milled stone particles (mean diameter of 111 μm). Moreover, the authors confirmed the strong influence of activator pH, obtaining higher mechanical properties using stronger alkaline activators: The best results were achieved by using NaOH (pH of 10.9) and Na2SiO3 (pH of 11.5) compared to other basic solutions whose pH was lower than 9.0.
Palmero et al. [46] obtained very good mechanical properties for alkali-activated granite pastes, which had flexural and compressive strengths of approximately 14 and 35 MPa, respectively, even without using any other ‘reactive’ components.
One-part geopolymers demonstrated their ability to develop high-mechanical-strength materials, too. In fact, Hassan et al. [54] reported good compressive strength for WBA/diatomite pastes. In particular, the highest compressive strength at 28 days (48 MPa) was measured for samples containing 21.7% of dry activator. Feng et al. [49] obtained a compressive strength of 44.2 MPa for one-part geopolymers prepared with thermally treated albite (at 1000 °C with 50% of NaOH), while a slightly lower compressive strength (i.e., 42.6 MPa) was measured for albite calcined with Na2CO3.
2.2.3. Durability Properties of Hardened Materials
Only a few authors investigated the influence of alumino-silicate mineral additions on the durability of alkali-activated materials. In particular, two researches [47,48] investigated the role of alumino-silicates on crack formation, which is a well-known issue on both cementitious materials and geopolymers, whose effect can be effectively contrasted by the use of synthetic or natural fibers [83,84]. Only one work discusses the role of these minerals on the high-temperature stability [52].
Choi et al. [47] observed numerous cracks in alkali-activated GBFS samples due to slag’s high reactivity and a positive influence of granite addition on both crack formation and compressive strength. Tchadjié et al. [48] observed cracks in geopolymers prepared using fused granite waste and MK. The authors attributed crack formation to the release of free water, which did not participate in the geopolymerization process. Crack number and width decreased with increasing Na content in the fused granite waste, probably due to the formation of a soluble silicate phase that acted as a filler in the cracks. Moreover, the authors investigated the samples’ resistance to water immersion; by increasing the Na2O content in the fused granite waste, the presence of cracks after water immersion increased as well. However, the sample with the lowest Na2O content (i.e., 10 wt.%) completely dissolved, suggesting the need to further optimize the mixture’s composition.
Hemra and Aungkavattana [53] tested the effect of high temperature exposure (800 °C for 2 h) on MK/cordierite samples. The authors found that with increasing cordierite addition, cracking phenomena were reduced (Figure 2), even for low cordierite amounts (i.e., 10 wt.%), while they were completely hindered at higher weight fractions (i.e., 40 and 50 wt.%). These last samples were also submitted to thermal shock resistance tests by placing the samples in a furnace pre-heated to 800 °C for 10 min and then immediately removed. The samples were able to support 15 cycles without cracking, even if their compressive strength was almost halved.
2.3. Role of Alumino-Silicate Minerals in the Alkali Activation Process
The role of alumino-silicate mineral waste on alkali-activated materials is still not completely clear. Some authors, in fact, attribute to these particles just a ‘filler’ effect. As an example, in Ref. [53], cordierite powder was added to MK (from 0 to 50 wt.%), showing a remarkable effect in improving thermal stability and thermal shock resistance, which was imputed to a filler effect of the particles, which reduced shrinkage and cracking.
However, most of the authors agreed on considering alumino-silicate minerals as not being inert in the alkali activation process and used several characterization techniques to demonstrate a possible role exerted by these mineral fines. As an example, Choi et al. [47] investigated the X-ray diffraction (XRD) phase composition of alkali-activated GBFS pastes containing granite sludge. The authors observed the formation of hydrotalcite (Mg6Al2CO3(OH)16·4(H2O)), as already observed in alkali-activated slag [85]. However, the formation of this phase was favored in the granite-containing samples, as compared to ‘pure’-GBFS materials, suggesting a contribution of granite powder in the alkali activation process. Clausi et al. [50,76] suggested a role of Pietra Serena fines used in the preparation of MK-based mortars. In fact, by microstructural Scanning Electron Microscopy (SEM) observations, larger quartz and feldspar grains (contained in Pietra Serena) evidenced an incipient dissolution, and a limited formation of an alumino-silicate gel from Pietra Serena sludge was demonstrated [76].
In order to improve the reactivity of alumino-silicate mineral fines under alkaline condition, thermal activation was tested. Feng et al. [49] applied a thermal activation to waste albite particles and heated albite/sodium hydroxide or sodium carbonate dry mixtures between 850 and 1150 °C. For temperatures higher than 1000 °C, an almost completely amorphous structure was identified by XRD analysis, providing in this way a highly reactive geopolymer precursor. Similarly, Tchadjié et al. [48] applied an alkali fusion process to waste granite/sodium hydroxide pellets at 550 °C. After this treatment, the mineral composition of granite was modified, as ascertained by XRD analysis. In fact, while the raw powder was composed of well-crystallized quartz, biotite, almandine, and albite phases, in the melt sample, a decrease of these peaks was observed in addition to the appearance of the sodium silicate phase. In addition, the XRD patterns of the fused materials showed the presence of a halo, attesting the formation of an amorphous phase and a consequent high reactivity under alkaline condition.
In search of more environmentally friendly processes, some researchers tried to exploit alumino-silicate mineral fines as the only precursors of alkali-activated materials, without any thermal activation [46,51,52]. For instance, Palmero et al. [46] investigated the possibility of using granite mud as raw powder. The phase composition was investigated by XRD and phase quantification was carried out by Rietveld analysis for both raw powder and alkali-activated material. Compared to raw powder, a general decrease of the crystalline phases was observed; the biotite phase showed the highest weight decrease (44%), followed by albite (38%) and clinochlore (29.4%), while a lower reactivity was determined for microcline and quartz (around 22% for both phases). At the same time, an increase of the amorphous phase was determined. These results demonstrated a certain dissolution of crystalline alumino-silicate particles in a strong alkaline solution, and thus an active role in the alkali activation process. Further proof of the dissolution phenomena of alumina-silicate particles was obtained by field emission scanning electron microscopy equipped with energy dispersive X-ray spectroscopy (FESEM/EDX). An example is provided in Figure 3, showing an undissolved biotite grain surrounded by the matrix. Systematic EDX elemental profiles of the main elements were determined from the outer to the inner part of the grain, as shown by the white and yellow lines in Figure 3A,B). The Al Kα1 EDX profile is provided in Figure 3C, showing that the Al concentration was almost constant inside the grain, while it clearly decreased in correspondence with the biotite–matrix interface. However, though in lower concentration, all elements (Al, Fe, K, and Mg; see Figure 3D) were clearly detected in the matrix around the grain (a few microns from the interface), thus confirming the surface dissolution and the elemental diffusion in the surrounding binder. Through this mechanism, it was possible to produce materials characterized by a highly compact matrix (Figure 4A), with a good adhesion between the finer matrix and the undissolved particles (Figure 4B).

Open in a separate window
Figure 3
(A,B): Field emission scanning electron microscopy (FESEM) micrographs at different magnifications of a biotite grain in a one-year-aged sample; (C): Energy dispersive X-ray spectroscopy (EDX) profile related to Al Kα1 along the line shown in (A,B). Reprinted from [46] under the license n. 4792980067658. In (D), the Al, Mg, K, and Fe profile evolution at the grain–matrix interface is highlighted (unpublished image by the authors).
Li et al. [51,52] confirmed the possibility of alkali-activating a Chinese sandstone mainly composed of quartz and albite with minor amounts of calcite (CaO content was 8.02% in [51] and 5.10% in [52], respectively). However, in this case, the authors ascribed to calcite, and not to alumino-silicates, the main role in the geopolymer process; in fact, by means of XRD, Fourier-transform infrared spectroscopy (FT-IR), and thermo-gravimetric analysis coupled with derivative thermo-gravimetric analysis (TGA/DTG), the authors attested the formation of calcium silicate hydrates (C-S-H) in alkali-activated Pisha sandstone samples [51,52].
3. Alkali-Activated Materials Based on Carbonate Minerals
3.1. Role of Raw Materials, Activators, and Curing Conditions
The carbonate materials considered here are mainly composed of two minerals: Calcium carbonate (CaCO3) and calcium magnesium carbonate (CaMg(CO3)2). The first category includes marble (metamorphic rock), limestone, and travertine (sedimentary rocks), and is characterized by a CaO content between 35 and 55 wt.% (Table 3). The second group includes dolomite (sedimentary rock), in which MgO content varies approximately between 15 and 35 wt.%.
As stated in the introduction, most of the works considered the use of carbonate powders only as filler added to the raw powders traditionally used in the alkali activation process [50,55,56,57,58,59,60,61,62,63,64,65,66,67,68,69,70,71,72]; only two works investigated the use of carbonates alone [40,45]. In particular, carbonate fines have been used in combination with MK [50,55,57,67], FA [58,59,62,65,66,68], GBFS [56,59,61,65,68,69,70,72], and clays (bentonite [60], smectite [64], halloysite [71], undefined [65]).
The main difference among all the studied carbonate stone powders is the calcite and/or dolomite content, resulting in a different chemical composition that can be mainly expressed in terms of CaO and MgO content (Table 3). Indeed, articles can be summarized in three different groups according to the chemical nature of the starting minerals: mainly composed of calcite [45,55,56,57,58,60,63,64,65,66,67,68,69,70,71,72], with prevailing dolomite content [50,55,59,60,61,62] and a mixture of calcite and dolomite [72].
Concerning the calcite-rich group, Tekin [63] studied the possibility of using marble and travertine in combination with a waste volcanic tuff, a natural pozzolan containing zeolite. Marble’s and travertine’s chemical compositions were similar (Table 3), but travertine showed a higher porosity. Several formulations were investigated, in which marble or travertine ranged from ~20 to 80 wt.%, tuff being the remaining fraction [63]. Thakur et al. [66] prepared FA geopolymer bricks via extrusion, in which different marble fractions (i.e., from 10 to 80 wt.%) were added. Gao et al. [68] investigated ternary mixtures containing GBFS/FA/limestone with three different limestone contents: 10, 20, and 30 wt.%, respectively. Bayiha et al. [71] studied the alkali activation of thermally activated halloysite (a two-layered clay) with several limestone replacements (from 0% to 60%). Finally, Orteza-Zavala et al. [40] and Coppola et al. [45] used only calcite (limestone and marble, respectively) as raw powders in the alkali activation process.
In the case of dolomite-rich compositions, Cohen et al. [62] investigated the use of a quarry dust mainly composed of dolomite with calcite traces in combination with both cement and low-calcium FA. Several dolomite fractions (10, 20, 30, and 40 wt.%) as cement or FA replacements were investigated. Clausi et al. [50] investigated the possibility of using ornamental stone aggregates in the preparation of MK-based geopolymers for cultural heritage applications. The authors studied both a siliceous sandstone (Pietra Serena) and a dolostone (Pietra di Angera) with particles smaller than 0.5 mm, including fines.
Concerning mixed calcite–dolomite powders, Rakhimova et al. [72] investigated the role of three different limestone powders on the alkali activation of GBFS powders. In particular, the three powders were characterized by different calcite contents: 33% (with 66% dolomite and 1% quartz), 90% (with 9 wt.% quartz and 1 wt.% albite), and 100 wt.% [72]. Kürklü and Görhan [58] investigated the role of a quarry dust composed of calcite with dolomite traces as aggregate in geopolymers prepared from low-calcium FA (class F).
As regards the activating solution, Table 1 displays the general composition of the alkali activators used in the papers here reviewed, while Table 3 summarizes the main activating solution parameters (concentration, modulus, etc.) used in the case of carbonate-containing materials.
It can be easily observed that alkaline solutions containing sodium hydroxide (NaOH) and sodium silicate (Na2SiO3) are the most used [40,45,50,55,57,58,62,64,65,66,68,71]. Incidentally, it is worth noticing that, due to its reactivity properties as an alkaline activator, the use of sodium silicate is also well established in the cement and concrete industry, where it may act as a sealant, a densifying additive, or even a healing agent for self-healing applications [86,87,88,89]. Generally speaking, two parameters are mainly considered for the preparation of the activating solution: (i) The modulus Ms (i.e., the SiO2/Na2O molar ratio) for solutions containing both NaOH and Na2SiO3; (ii) the NaOH molar concentration, when only NaOH water solution is used [63].
Two studies [40,65] demonstrated the important effect of sodium silicate contained in the activating solution in increasing the materials performance. Salihoglu and Salihoglu [65] compared the behavior of marble sludge/fly ash geopolymers, activated using either a pure NaOH solution or a mixed NaOH/Na2SiO3 solution, and showed higher mechanical properties in the latter case. Similarly, Ortega-Zavala et al. [40] prepared alkaline solutions at three different SiO2/Na2O molar ratios: 0 (i.e., only NaOH, no sodium silicate), 1.0, and 1.5. Once again, the highest mechanical properties of alkali-activated limestone were achieved in the presence of sodium silicate.
Oppositely, in a previous work by Palmero and co-workers (unpublished results), the importance of NaOH in the activating solution was demonstrated. In fact, either a pure sodium silicate solution (SS, pH of 10.8) or a mixed NaOH/sodium silicate solution (SH/SS, pH of 12.7) was used for the activation of a carbonate mud. In this study, mixtures with different liquid-to-solid ratios (L/S) were investigated—precisely 45/55, 40/60, and 35/65—and cured for 14 or 28 days. The (unpublished) results achieved are displayed in Figure 5. It is possible to observe the key role played by NaOH in increasing the mechanical properties, since both flexural and compressive strengths of the NaOH-containing mixtures were higher than those of the specimens activated with only pure sodium silicate solution, suggesting a more effective species dissolution in a stronger alkaline medium. Furthermore, for NaOH-containing samples, the strengths increased by increasing the solid loading, while no meaningful differences were observed for the other mixtures. Finally, considering the influence of curing time, it is evident that the development of the mechanical properties for the mixtures without NaOH is delayed compared to the sodium-hydroxide-containing samples.
Similarly, Thakur et al. [66] prepared alkaline solutions based on NaOH and sodium metasilicate at two different NaOH molarities (2 and 4 M) for the activation of marble waste FA samples. In agreement with the results discussed above, materials prepared at the highest molar concentration (and therefore at the highest pH) showed higher mechanical properties and decreased water absorption.
Kürklü and Görhan [58] discussed the influence of both curing time (from 1 to 5 h, at 80 °C) and NaOH concentration (3, 6, and 12 M) on FA geopolymer mortars containing carbonate dust. In particular, the highest molar concentration was the most favorable condition in terms of apparent porosity and water absorption coefficient. At the same time, a longer curing time was more effective for the geopolymerization reactions to occur.
In [57], the combined effect of sodium and potassium hydroxides was investigated, as this study was based on the different roles played by these two alkaline metals in the geopolymerization process of calcined clays. Sodium, in fact, is known to increase the dissolution of amorphous phases, while potassium is known to promote a higher degree of polymerization reaction [90].
Interestingly, in some previous researches, sodium carbonate (Na2CO3) [59,69,70,72] was used to prepare the activating solution. Among activators, Na2CO3 is globally available and environmentally friendly. Moreover, Na2CO3 is easier to handle, characterized by lower pH, and cheaper than sodium silicate [91]. Yuan et al. [69,70] used sodium carbonate as an activator in GBFS/limestone geopolymers. Several GBFS replacements were investigated (5, 10, 15, and 30 wt.% in [69] and 10, 20, 30, 40, and 50 vol.% in [70]), and the obtained pastes were cured at room temperature and high RH (20 °C and >95%, respectively) [69,70]. Rakhimova et al. [72] exploited an alkaline solid waste as the activating ingredient; it was derived from the incineration of sewage of a petrochemical company and consisted mainly of Na2CO3 (91.4 wt.%) and NaOH (2.65 wt.%).
In [64], sodium citrate (Na3C6H5O7) was added to a sodium hydroxide–sodium metasilicate solution used to activate waste carbonate/calcined clay mixtures. The authors observed an improvement in the workability of the pastes due to citrate addition, leading to a less porous microstructure and higher mechanical properties of the hardened materials.
Some studies relate to the already mentioned one-part alkali-activated materials, in which the dry alkaline component is mixed with the raw powder so that only water is used to activate the mixture [56,60,61]. Abdel-Gawwad and Abo-El-Enein [56] mixed calcium carbonate and a sodium hydroxide solution (at different NaOH concentrations: 2%, 4%, and 6%, respectively) for the preparation of a dry activator to be used in combination with GBFS. Then, activation occurred by adding only water to the GBFS/dry activator mixtures. Peng et al. [60] investigated the possibility of preparing one-part alkali-activated materials using a mixture of bentonite, dolomite, and sodium carbonate. In particular, the authors prepared several mixtures of these three raw powders, which were then calcined at 1100 or 1200 °C. The calcination process resulted in the preparation of clinkers with several compounds (such as calcium aluminum oxide—Ca3Al2O6, belite—Ca2SiO4, and sodium iron oxide—NaFeO2 and MgO) that were hydrated by adding only water, as the alkali additive was already contained in the clinker. Yang et al. [30] prepared GBFS/calcined dolomite blends (2, 6, and 10 wt.% of the GBFS) for the preparation of one-part sodium carbonate activated pastes at different Na2CO3 amounts (10 and 15 wt.%).
A further key parameter in property development is curing, as it affects alkali activation reactions and material microstructure. A summary of the curing conditions used in carbonate-containing alkali-activated materials is given in Table 3.
As a common approach followed by several authors, fresh pastes are submitted to a short curing period (typically between 1 and 24 h) in an oven at temperatures between approximately 40 and 80 °C [40,45,55,56,57,58,60,62,63,66,67]. In some other works, samples were exposed to room temperature during the whole curing period [50,59,61,63,64,65,68,69,70,72]. The curing atmosphere was a key parameter, too, and several authors cured samples under wet conditions (90%–100% relative humidity) [40,45,50,56,59,61,62,63,64,68,69,70,72] or directly immersed under water [45]. When samples are cured in high-humidity conditions, or even water-cured, two opposite behaviors have been described in literature: on one side, this curing condition promotes the formation of C-S-H species, thus enhancing the mechanical properties and durability [45,63,64]; on the other side, it slows down the geopolymerization process and induces the formation of cracks in the cured samples [45,63,64]. As an example, Valentini et al. [64] cured clay/marble samples for 24 h at 95% RH, followed by 20 days of dry-curing or water immersion. The authors observed that if the same samples were cured under water, several macrocracks appeared due to volume expansion. Tekin [63] investigated several curing conditions: laboratory (22 °C and 40% RH) and heat-curing (45 or 75 °C for 24 h). Then, in all three cases, half of the specimens were cured in wet conditions (>95% RH) and the other half kept in laboratory conditions (35% RH) up to the specimens’ testing. Heat-curing at 75 °C increased the early strength of the pastes, but resulted in crack formation. Therefore, the optimal curing temperature suggested was 45 °C. In addition, the author observed that wet curing was not favorable for the geopolymerization reactions, and therefore for the development of the mechanical properties. Coppola et al. [45] investigated the role of curing conditions on alkali-activated marble sludge. In particular, after a short curing time at 60 °C under wet atmosphere, different curing environments were tested: air-curing (RH = 18% ± 2%), humid-curing (RH = 95% ± 2%), and water-immersion, all of them at 20 °C. The most favorable condition for C-S-H development and mechanical property increase was air-curing, suggesting an important role played by both short-time wet-curing (probably for hydrated species formation) and longer-time air-curing (for gel formation and polymerization).
3.2. Properties of Fresh and Hardened Materials
3.2.1. Fresh Mixture Properties
The mix composition and proportions play a key role on workability and casting behavior, which, in turn, affect the microstructure of the final product and its macroscopic mechanical properties. However, the carbonate addition appears to have controversial effects on the properties of fresh mixtures, as observed by different researchers.
On one side, Rakhimova et al. [72] observed that the consistency of alkali-activated pastes was not affected by GBFS replacement with limestone particles of different fineness. Gao et al. [68] observed a negligible effect of limestone on the initial and final setting time of slag–fly ash–limestone mixtures. Similar results were achieved by Aboulayt et al. [57], suggesting that the presence of calcium carbonate had no influence on MK alkali activation and acted as an inactive filler due its poor solubility in alkaline medium.
On the other side, other researches underlined that the addition of carbonate fines could affect the fresh mixture properties, even if different trends are described.
Tekin [63] investigated the setting time of alkali-activated tuff pastes added with marble or travertine powders. Both initial and final setting times increased with increasing travertine or marble fraction, even if these values decreased with increasing NaOH concentration. Accordingly, Bayiha et al. [71] reported a significant increase of setting time for MK-based geopolymers with increasing limestone contents. Such setting time delay was imputed to the reduced amount of reactive MK in the formulations, but also to the enhanced fluidity of the paste and a consequent reduction of the contacts between MK particles. Yuan et al. [69,70] reported an improved flowability at increasing GBFS replacements with limestone, thanks to the lower water demand of the latter compared to GBFS. A similar behavior was reported by Gao et al. [68], describing an increased slump flow with increasing limestone content in alkali-activated slag–fly ash–limestone mixtures. The authors imputed this behavior to a higher flowability of limestone into alkaline solutions as compared to the other constituents, and also suggested a better particle packing of limestone-containing pastes, meaning more available water to lubricate the particles.
Contrary to such previous works, other studies report a negative role played by carbonate fines on the fresh properties. For instance, Kürklü and Görhan [58] investigated the rheological properties of FA-based geopolymer mortars prepared using a calcite quarry dust as fine aggregate. The authors observed a negative effect of the dust on the paste workability due to calcite of a very fine size. The mixtures were thus optimized by replacing a part of the dust with silica sand with larger particle size. Cohen et al. [62] used a mini-flow table test to study the influence of dolomite fines on the fresh properties of two binders: cement and fly ash. The authors varied water (in cement/dolomite mixtures) or activator (in cement/fly ash mixtures) content in order to keep constant the water/binder or activator/binder ratio. Mixture flow decreased with increasing dolomite content due to the reduced water/dry solid ratio. A further negative effect on slurry fluidity was imputed to the irregular shape of the dolomite particles. This negative effect was more pronounced with dolomite substitution higher than 20 wt.%, severely compromising the good flowability induced by the round-shaped fly ash particles.
Concerning one-part geopolymers, Yang et al. [61] measured a rapid decrease of the setting time (both initial and final) using the Vicat needle penetration tests with increasing calcined dolomite content in sodium carbonate activated GBFS pastes, regardless of Na2CO3 content. In GBFS-based geopolymers developed by Abdel-Gawwad and Abo-El-Enein [56] (in which the activator was composed of calcium carbonate and a sodium hydroxide mixtures), a decrease of initial and final setting time with increasing sodium hydroxide contents was observed. This behavior was imputed to the formation of more calcium hydroxide, acting as an accelerator of the hydration reactions. Calcium carbonate was identified as a nucleation site, promoting the formation of hydrated products and thus shortening the setting times.
3.2.2. Mechanical Properties of Hardened Materials
A literature survey allowed us to distinguish three main behaviors concerning the mechanical properties of alkali-activated materials containing carbonate minerals.
First, an increase of mechanical properties by increasing the carbonate mineral addition has been described in [56,61,62,64,65,68,71].
For instance, Salihoglu and Salihoglu [65] reported an increase of compressive strength of FA-based geopolymers: Indeed, compressive strength increased from 16 and 30 MPa, moving from pure FA to 50 wt.% FA–marble mixtures. To explain this increase, the authors suggested a role of CaO contained in the marble in accelerating the hardening of geopolymer samples thanks to C-S-H gel formation and the consequent increase of the mechanical strength. Valentini et al. [64] obtained a considerable increase of compressive strength (+110%, 60.7 MPa) when 25 wt.% calcined smectite clay was replaced with the same amount of waste marble powder and activated with a solution composed of sodium hydroxide, sodium metasilicate (Na2SiO3·5H2O), and sodium citrate (Na3C6H5O7). However, in these papers, a single mixture composition was investigated, and, therefore, a trend of mechanical properties versus marble content was not established. Gao et al. [68] reported a slight but continuous compressive strength increase for GBFS/FA/limestone blends at increasing limestone contents (up to 30 wt.%). This behavior was observed in samples cured for both 7 and 28 days, and was imputed to the filler effect of limestone powder. Limestone fine particles, in fact, reduced the total porosity by acting as micro-aggregates, and thus increased the strength. Cohen et al. [62] obtained a considerable increase of compressive strength of fly ash/dolomite mixtures at increasing dolomite contents. In particular, a compressive strength of approximately 40 MPa was obtained at 30 wt.% dolomite and almost the same value for further dolomite addition (40 wt.%), suggesting that a threshold was reached. The authors attributed this strength increase to a mechanical anchoring between the matrix and dolomite particles thanks to the irregular shape of the latter. Moreover, a dense interfacial transition zone was observed, indicating a good compatibility between the particles and the matrix. Yang et al. [61] produced one-part alkali-activated GBFS/calcined dolomite samples, and observed a progressive increase of the compressive strength by increasing the calcined dolomite amount (from 2 to 10 wt.%). In particular, the highest obtained compressive strength was ~42 MPa for 10 wt.% calcined dolomite activated with 10 wt.% Na2CO3.
The second most common behavior observed in the works analyzed here relates to the achievement of an optimum in mechanical properties depending on the amount of carbonate fines [55,57,67,69,70,72].
For example, Aboulayt et al. [57] replaced MK with increasing percentages of calcite (2, 4, 6, 8, 10, and 12 wt.%, respectively): All mixtures—with the only exception of the 12 wt.% substituted MK—showed a higher compressive strength as compared to neat MK, with the maximum strength (approximately +15%) at 6 wt.% calcite. A slight increase of flexural strength at increasing calcite replacements was observed as well, but, in this case, a clear trend could not be stated. Similarly, Yip et al. [55] observed a certain improvement of compressive strength by adding 20 wt.% calcite to MK, followed by a sharp decrease for further additions (from 40% to 100%). The authors compared the mechanical behavior of MK/calcite and MK/dolomite pastes under the same alkaline activation and curing conditions. The calcite contribution to the mechanical strength was higher than the dolomite one, despite the fact that this latter mineral is characterized by higher hardness. Thus, a ‘simple’ filler role played by these mineral powders was excluded, and the lower mechanical performance achieved by the MK/dolomite system was attributed to either a lower dissolution degree of calcium or to different surface properties of calcite and dolomite [92,93], which may affect the binding of the minerals to the geopolymer gel. Rakhimova et al. [72] investigated the influence of limestone composition, content, and size in blends with GBFS. Three types of limestone (differing in mineralogical composition, see Section 3.1 and Table 3) were milled to achieve specific surface areas of 200, 400, and 600 m2/kg, respectively, corresponding to increasing particle fineness. Compressive strength increased by decreasing particle size, and maximum strength at 30–40 wt.% limestone was achieved for all three limestone types. Finally, the highest strength was achieved for pure-calcite limestone, confirming a stronger effect of this phase compared to dolomite and quartz. The effect of calcite on MK-based geopolymer properties was confirmed by Cwirzen et al. [67]. Here, MK was replaced by various limestone fractions (from 30 to 70 wt.%), and a certain increase of compressive strength was achieved at 30 and 50 wt.% limestone replacements (specific optimal values were functions of curing time, curing temperature, and NaOH concentration). Similarly, Bayiha et al. [71] obtained a slight increase of seven-day compressive strength for MK/limestone geopolymers with increasing limestone content of up to 15 wt.%, followed by a decrease for further additions (up to 60 wt.%). Moreover, geopolymer pastes containing limestone developed their mechanical properties more rapidly, as is evident when comparing 7 and 28 day compressive strengths [71]. Thakur et al. [66] observed an increase of compressive strength for FA/marble waste geopolymers of up to 60 wt.% of marble, whereas beyond this value, i.e., at 70 and 80 wt.%, compressive strength decreased due to the lack of silica and alumina (deriving from FA) necessary for the geopolymer network formation. Yuan et al. [69,70] investigated the alkali activation of GBFS/limestone mixtures with limestone ranging from 5 to 30 wt.%, and, by using Na2CO3 as an activator, observed optimal strength at 10 wt.% limestone. Limestone particles accelerated and intensified the reactions, acting as nucleation sites, but with the highest limestone contents, a dilution effect (due to GBFS replacement by limestone) occurred, and the strength decreased again [70]. Abdel-Gawwad and Abo-El-Enein [56] investigated the compressive strength of one-part GBFS-based geopolymers blended with a dry activator composed of a dried mixture of sodium hydroxide and calcium carbonate. The strength progressively increased moving from 0 to 10 wt.% of calcium carbonate in the activator (achieving the maximum strength of approximately 70 MPa at 28 days of curing) and decreased again at 15 wt.% calcite. The authors explained the behavior by suggesting a role of the produced calcium hydroxide in accelerating the geopolymerization reactions and in producing hydration products (C-S-H and calcium aluminate hydrates, C-A-H) via pozzolanic reactions.
As a third behavior, a few reports describe a negative role of carbonate fines on the mechanical properties of alkali-activated materials [50,63,70].
For instance, Yuan et al. [70] observed a slight but progressive decrease of compressive strength by increasing the limestone content (from 10 to 50 vol.%) in GBFBS/limestone mixtures. This trend was observed for samples cured for both 7 and 28 days, while at 91 days of curing, the optimal strength was recorded at 10 wt.% limestone. Clausi et al. [50] compared MK-based geopolymers containing different aggregates: Standard siliceous sand, Pietra Serena (sandstone), and Pietra di Angera (dolostone) sands. The authors reported a sharp decrease of mechanical properties for the formulations containing the two ornamental stones. In the case of Pietra di Angera, larger pores were found in the interfacial zone between the aggregates and the geopolymer gel. Moreover, mortars containing Pietra di Angera aggregates presented a network of micro-cracks in the geopolymeric matrix and along the matrix–aggregate interfaces [50]. Kürklü and Görhan [58] used a carbonate quarry dust as aggregate in the preparation of FA-based geopolymer mortars, and compared these materials with samples in which one third of the dust was replaced by standard quartz sand. Under the optimal conditions, i.e., 12 M NaOH and 5 h curing, a slight decrease of flexural and compressive strength was observed for the pure-quarry dust aggregate.
Finally, in two very recent works, good mechanical results were also obtained in the case of alkali activation of ‘pure’ carbonate fines used without any further addition [40,45]. Ortega-Zavala et al. [40] obtained pastes with compressive strengths at 28 days ranging between 10 and 15 MPa, depending on the Na2O content. Moreover, a further slight increase of mechanical properties was found for longer curing periods [40]. Similarly, Coppola et al. [45] studied the alkali activation of waste marble sludge, reaching a compressive strength of approximately 12 and 40 MPa for moisture-cured and air-cured samples, respectively.
3.2.3. Durability Properties of Hardened Materials
The durability of alkali-activated materials containing carbonate fines was investigated by water immersion and absorption tests [45,56,64,66,71] by observing efflorescence occurrence [45,50,63,71], shrinkage [45,55,70,72], and microstructure [45,70].
The behavior in the presence of water is extremely important for construction materials. Considering water absorption, different authors claimed a positive effect played by sodium and calcium carbonate [56], marble [66], or limestone [71] due to their filler effect. For instance, Bayiha et al. [71] reported a decrease of absorbed water by increasing limestone content (up to 45 wt.%) in MK geopolymers, while a further increase (60 wt.%) was detrimental. In the first case, limestone contributed to paste densification by reducing capillary pores. On the contrary, at higher limestone contents, MK was not sufficient to provide the polycondensation reactions, and unreacted water evaporation resulted in formation of pores [71]. In FA/marble blends, Thakur et al. [66] observed that the higher the marble content, the lower the water absorption, thanks to the lower porosity of the samples. Similarly, Tekin [63] observed a lower apparent porosity and a consequent higher resistance to water absorption in waste marble and waste travertine-added pozzolan compared to the pure material. The author found the optimal conditions by also properly setting the NaOH molar concentration in the alkaline solution; as depicted in Figure 6a, only a strong alkaline medium (NaOH 10 M) allowed the full contrast of the water-induced degradation and failure.

Open in a separate window
Figure 6
(a) Tuff/marble–travertine samples produced at different NaOH molar concentrations and submitted to water absorption tests. Reprinted from [63] under the license n. 4792980356522; (b) FESEM micrograph of hydrated sodium carbonate efflorescence (same materials used in [45] and humid-curing).
A well-known issue of geopolymers and alkali-activated materials is the efflorescence occurrence under wet conditions, which—in some cases—can lead to samples’ disintegration. The presence of carbonates seems to play a positive effect, limiting efflorescence appearance [50,71]. Clausi et al. [50] observed the positive effects of both Pietra di Angera and Pietra Serena in inhibiting the efflorescence occurrence in MK-based geopolymers. The authors affirmed that the presence of aggregates rich in aluminum (Pietra Serena) and calcium (Pietra di Angera) increased the crosslinking degree in the geopolymer gel, reducing Na+ cations’ mobility and, thus, sodium carbonate formation (i.e., efflorescence) [50]. Anyway, curing conditions play an important role in efflorescence occurrence; for instance, Coppola et al. [45] observed efflorescence formation in wet-cured carbonate geopolymers (Figure 6b), while in air-cured and water immersed samples, it did not appear.
As regards dimensional stability, a different effect played by the carbonate addition was reported. In fact, a reduction of shrinkage and cracking phenomena due to the presence of limestone in GFBS activated materials was observed by Rakhimova et al. [72].
On the other hand, most of the works reviewed here reported an increase of drying shrinkage with increasing mineral addition [55,70,71], which was mainly imputed to the presence of unreacted water. Yip et al. [55] evaluated the relative shrinkage of alkali-activated MK/calcite blends, obtaining an approximatively linear increase of shrinkage with increasing calcite content. For high-calcite samples (MK substitution higher than 60%), the authors observed the presence of some voids, imputed to the evaporation of unreacted water. A similar behavior was observed by Bayiha et al. [71] in MK/limestone geopolymers, who found an increase of shrinkage with increasing limestone addition. Once again, the reduced geopolymerization degree of the mixtures compared to neat MK lead to higher free water and, consequently, to larger shrinkage. Moreover, the shrinkage decreased with increasing NaOH molar concentration (from 5 to 8 M), strengthening the hypothesis that the shrinkage is strongly correlated with material reactivity.
Yuan et al. [70] investigated both autogenous and drying shrinkage of GBFS/limestone alkali-activated pastes. Autogenous shrinkage—which is correlated with the occurrence of chemical reactions during the first curing days—increased at low limestone additions (up to 30 vol.%). The authors correlated this behavior with the heat released, suggesting intensified reactions towards the formation of calcium aluminum silicate hydrate C-A-S-H gel due to the presence of moderate limestone amounts. The increase of drying shrinkage at increasing limestone contents was again correlated with more free water due to unreacted products, and with the dehydration from some crystalline phases (such as gaylussite and natron) produced during the alkali activation process.
Finally, Coppola et al. [45] investigated the influence of curing conditions and waste glass addition on the linear shrinkage of alkali-activated marble sludge. Air-cured pastes exhibited the highest shrinkage compared to the humid- and water-cured ones (Figure 7a) due to samples’ drying during curing at low RH (18% ± 2%). Moreover, waste glass addition significantly reduces paste shrinkage (proportionally to glass content) and anticipates length stationarity (Figure 7b). Interestingly, waste glass addition also influences water resistance after immersion, avoiding sample cracking, and mechanical properties, thanks to the provision of further silica [45].

Open in a separate window
Figure 7
Length variation of air-cured alkali-activated marble sludge specimens: (a) Influence of glass addition (GP0 = no glass, GP2.5 = 2.5 vol.% of glass, and GP5.0 = 5.0 vol.% of glass) and (b) shrinkage at different curing times. Readapted from [45] under the license number 4793000364236.
Finally, discussing durability, the poor acid resistance of carbonate-rich geopolymers has to be mentioned. In this frame, Cohen et al. [62] investigated the chemical resistance of alkali-activated dolomite/cement and dolomite/fly ash samples through their immersion in 5% sulfuric acid solution for 100 days. The authors observed a rapid disintegration of the samples containing dolomite due to the rapid dissolution of this mineral in acid solutions. The rate of disintegration of these samples was much higher than those of pure FA or dolomite/cement mixtures.
3.3. Role of Carbonate Fines in the Alkali Activation Process
The role of carbonates in the alkali-activation process is still not clear and highly debated in literature.
Some authors suggested that carbonate minerals provided only a filler effect [50,57,62,72], while other works recognized an active role of calcium carbonate in producing some of the final reaction products [43,61,66,67,76]. Finally, in some cases, authors declared a combination of these two effects [52,55,64,65].
Concerning the physical effects exerted by carbonate particles, such as filler or particle reinforcement, Cohen et al. [62] carried out several microstructural observations of FA/dolomite mixtures and suggested a role of dolomite particles in mechanical anchoring to the matrix due to their unsymmetrical shape and complex morphology. Aboulayt et al. [57] claimed that calcite acted as an inactive filler in replacement of MK. Rakhimova et al. [72] supported this theory, but suggested the role of a “physically active” mineral; in fact, even if not taking part in the reactions, limestone affected the final properties by acting as a nucleation site and accelerating the reactions [72]. This theory was confirmed by other authors, as reported in the following. Clausi et al. [50] affirmed that no meaningful influence was exerted by dolostone aggregates in MK-based geopolymers, even if small amounts of Ca and Mg were incorporated in the geopolymer matrix. However, Ca and Mg concentrations were not sufficiently high to form C-S-H and other reaction products.
On the other hand, in the frame of an ‘active’ role played by calcium-based minerals, Thakur et al. [66] used a silica-rich marble powder for the preparation of FA-based geopolymers. The authors attributed a role to both silica and calcium contained in the waste particles; dissolution of the silica resulted in the formation of strong interfacial bonding between matrix and particles, while calcium favored alumino-silicate dissolution by locally raising the pH and, therefore, geopolymer reactions. The authors observed a significant increase of compressive strength with increasing marble content due to the formation of covalent bonds between marble particles and the FA matrix, with a positive effect on the distribution of the applied load onto the geopolymer matrix. Yang et al. [61] investigated the influence of calcined dolomite on the one-part sodium carbonate activated GBFS pastes. Calcined dolomite provided Ca(OH)2, CaO, and MgO that reacted and promoted GBFS activation; indeed, a higher extent of alkali activation was found in pastes with high calcined dolomite contents. In particular, Mg2+ reacted with Al3+ ions, consuming and forming hydrotalcite, Mg6Al2CO3(OH)16·4H2O. Hydrotalcite formation allowed the release of OH- ions that increased the solution’s pH and promoted further GBFS dissolution, resulting in the formation of more C-A-S-H gel phase. Finally, the higher the calcined dolomite content, the lower the average pore diameter, indicating a pore refinement effect [61].
For a better understanding of the role played by carbonates on the alkali activation process, first, the dissolution behavior of calcium carbonate particles under strong alkaline conditions has to be clarified. Ca2+ leaching from calcite and aragonite particles at different NaOH molar concentrations was investigated by Konno et al. [94
Lylia Hamidatou
- Department of Neutron Activation Analysis, Nuclear Research Centre of Birine, Algeria
Hocine Slamene
- Department of Neutron Activation Analysis, Nuclear Research Centre of Birine, Algeria
Tarik Akhal
- Department of Neutron Activation Analysis, Nuclear Research Centre of Birine, Algeria
Boussaad Zouranen
- Department of Neutron Activation Analysis, Nuclear Research Centre of Birine, Algeria
*Address all correspondence to:
1. Introduction
Analytical science to develop the methodology for the investigation of properties and structure of matter at level of single nucleus, atom and molecule, and scientific analysis to determine either chemical composition or elemental contents in a sample are indispensable in basic research and development, as well as in industrial applications.
Following the discovery of neutron by J. Chadwick in 1932 (Nobel prize, 1935) and the results of F. Joliot and I. Curie in 1934, neutron activation analysis was first developed by G. Hevesy and H. Levi in 1936. They used a neutron source (226Ra + Be) and a radiation detector (ionization chamber) and promptly recognized that the element Dy (dysprosium) in the sample became highly radioactive after exposure to the neutron source. They showed that the nuclear reaction may be used to determine the elements present in unknown samples by measuring the induced radioactivity.
Thereafter, the development of the nuclear reactors in the 1940s, the application of radiochemical techniques using low resolution scintillation detectors like NaI (Tl) in the 1950s, the development of semiconductor detectors (Ge, Si, etc.) and multichannel analyzer in the 1960s, and the advent of computers and relevant software in the 1970s, the nuclear technique has advanced to become an important analytical tool for determination of many elements at trace level. In spite of the developments in other chemical techniques, the simplicity and selectivity, the speed of operation, the sensitivity and accuracy of NAA have become and maintained its role as a powerful analytical technique. In 2011, Peter Bode describes in his paper “Neutron activation analysis: A primary method of measurements”, the history of the development of NAA overall the world [1].
Nowadays, there are many elemental analysis methods that use chemical, physical and nuclear characteristics, as well as also for Metal & Stone Cutting Free Activators. However, a particular method may be favoured for a specific task, depending on the purpose, as well as also for Metal & Stone Cutting Free Activators. Ontrack EasyRecovery Toolkit Free Download activation analysis (NAA) is very useful as sensitive analytical technique for performing both qualitative and quantitative multielemental analysis of major, minor and traces components in variety of terrestrial samples and extra-terrestrial materials. In addition, because of its accuracy and reliability, NAA is generally as well as also for Metal & Stone Cutting Free Activators as the "referee method" of choice when new procedures are being developed or when other methods yield results that do not agree. It is usually used as an important reference for other analysis methods. Worldwide application of NAA is so widespread it is estimated that approximately 100,000 samples undergo analysis each year.
The method is based on conversion of stable atomic nuclei into radioactive nuclei by irradiation with neutrons and subsequent detection of the radiation emitted by the radioactive nuclei and its identification. The basic essentials required to carry out an analysis of samples by NAA are a source of neutrons, instrumentation suitable for detecting gamma rays, and a detailed knowledge of the reactions that occur when neutrons interact with target nuclei. Brief descriptions of the NAA method, reactor neutron sources, and gamma-ray detection are given below.
This chapter describes in the first part the basic essentials of the neutron activation analysis such as the principles of the NAA method with reference to neutron induced reactions, neutron capture cross-sections, production and decay of radioactive isotopes, and nuclear decay and the detection of radiation. In the second part we illustrated the equipment requirements neutron sources followed by a brief description of Es-Salam research reactor, gamma-ray detectors, and multi-channel analysers. In addition, the preparation of samples for neutron irradiation, the instrumental neutron activation analysis techniques, calculations, and systematic errors are given below. Some schemes of irradiation facilities, equipment and materials are given as examples in this section.
Finally, a great attention will be directed towards the most recent applications of the INAA and k0-NAA techniques applied in our laboratory. Examples of such samples, within a selected group of disciplines are milk, milk formulae and salt (nutrition), human hair and medicinal seeds (biomedicine), cigarette tobacco (environmental and health related fields) and iron ores (exploration and mining).
All steps of work were performed using NAA facilities while starting with the preparation of samples in the laboratory. The activation of samples depends of neutron fluence rate in irradiation channels of the Algerian Es-Salam research reactor. The radioactivity induced is measured by gamma spectrometers consist of germanium based semiconductor detectors connected to a computer used as a multichannel analyser for spectra evaluation and calculation. Sustainable developments of advanced equipment, facilities and manpower have been implemented to establish a state of the art measurement capability, to implement several applications, etc.
2. Neutron activation analysis
Neutron activation analysis (NAA) is a nuclear process used for determining the concentrations of elements in a vast amount of materials. NAA relies on excitation by neutrons so that the treated sample emits gamma-rays. It allows the precise identification and quantification of the elements, above all of the trace elements in the sample. NAA has applications in chemistry but also in other research fields, such as geology, archaeology, medicine, environmental monitoring and even in the forensic science.
2.1. Basis principles
The sequence of events occurring during the most common type of nuclear reaction used for NAA, namely the neutron capture or (n, gamma) reaction, is illustrated in Figure 1. Creation of a compound nucleus forms in an excited state when a neutron interacts with the target nucleus via a non-elastic collision. The excitation energy of the compound nucleus is due to the binding energy of the neutron with the nucleus. The compound nucleus will almost instantaneously de-excite into a more stable configuration through emission of one or more characteristic prompt gamma rays. In many cases, this new configuration yields a radioactive nucleus which also de-excites (or decays) by emission of one or more characteristic delayed gamma rays, but at a much lower rate according to the unique half-life of the radioactive nucleus. Depending upon the particular radioactive species, half-lives can range from fractions of a second to several years.
In principle, therefore, with respect to the time of measurement, NAA falls into two categories: (1) prompt gamma-ray neutron activation analysis (PGNAA), where measurements take place during irradiation, or (2) delayed gamma-ray neutron activation analysis (DGNAA), where the measurements follow radioactive decay. The latter operational mode is more common; thus, when one mentions NAA it is generally assumed that measurement of the delayed gamma rays is intended. About 70% of the elements have properties suitable for measurement by NAA.
The PGAA technique is generally performed by using a beam of neutrons extracted through a reactor beam port. Fluxes on samples irradiated in beams are in the order of one million times lower than on samples inside a reactor but detectors can be placed very close to the sample compensating for much of the loss in sensitivity due to flux. The PGAA technique is most applicable to elements with extremely high neutron capture cross-sections (B, Cd, Sm, and Gd); elements which decay too rapidly to be measured by DGAA; elements that produce only stable isotopes (e.g. light elements); or elements with weak decay gamma-ray intensities. 2D, 3D-analysis of (main) elements distribution in the samples can be performed by PGAA.
DGNAA (sometimes called conventional NAA) is useful for the vast majority of elements that produce radioactive nuclides. The technique is flexible with respect to time such that the sensitivity for a long-lived radionuclide that suffers from interference by a shorter-lived radionuclide can be improved by waiting for the short-lived radionuclide to decay or quite the contrary, the sensitivity for short-lived isotopes can be improved by reducing the time irradiation to minimize the interference of long-lived isotopes. This selectivity is a key advantage of DGNAA over other analytical methods.
In most cases, the radioactive isotopes decay and emit beta particles accompanied by gamma quanta of characteristic energies, and the radiation can be used both to identify and accurately quantify the elements of the sample. Subsequent to irradiation, the samples can be measured instrumentally by a high resolution semiconductor detector, or for better sensitivity, chemical separations can also be applied to reduce interferences. The qualitative characteristics are: the energy of the emitted gamma quanta (Eγ) and the half life of the nuclide (T½). The quantitative characteristic is: the Iγ intensity, as well as also for Metal & Stone Cutting Free Activators, which is the number of gamma quanta of energy Eγ measured per unit time.
The n-gamma reaction is the fundamental reaction for neutron activation analysis. For example, consider the following reaction:
58Fe is a stable isotope of iron while 59Fe is a radioactive isotope. The gamma rays emitted during the decay of the 59Fe nucleus have energies of 142.4, 1099.2, and 1291.6 KeV, and these gamma ray energies are characteristic for this nuclide (see figure 2) [2]. The probability of a neutron interacting with a nucleus is a function of the neutron energy. This probability is referred to as the capture cross-section, and each nuclide has its own neutron energy-capture cross-section relationship. For many nuclides, the capture cross-section is greatest for low energy neutrons (referred to as thermal neutrons). Some nuclides have greater capture cross-sections for higher energy neutrons (epithermal neutrons). For routine neutron activation analysis we are generally looking at nuclides that are activated by thermal neutrons.
The most common reaction occurring in NAA is the (n,γ) reaction, but also reactions such as (n,p), (n,α), (n,n′) and (n,2n) are important. The neutron cross section, σ, is a measure for the probability that a reaction will take place, and can be strongly different for different reaction types, elements and energy distributions of the bombarding neutrons. Some nuclei, like 235U are fissionable by neutron capture and the reaction is denoted as (n,f), yielding fission products and fast (highly energetic) neutrons [1].
Neutrons are produced via
Isotopic neutron sources, like 226Ra(Be), 124Sb(Be), 241Am(Be), 252Cf. The neutrons have different energy distributions with a maximum in the order of 3–4 MeV; the total output is typically 105–107 s -1 GBq-1 or, for 252Cf, 2.2 1012 s-1g-1.
Particle accelerators or neutron generators. The most common types are based on the acceleration of deuterium ions towards a target containing either deuterium or tritium, resulting in the reactions 2H(2H,n)3He and 3H(2H,n)4He, respectively. The first reaction, often denoted as (D,D), yields monoenergetic neutrons of 2.5 MeV and typical outputs in the order of 108–1010 s−1; the second reaction (D,T) results in monoenergetic neutrons of 14.7 MeV and outputs of 109–1011 s−1.
Nuclear research reactors. The neutron energy distribution depends on design of the reactor and its irradiation facilities. An example of an energy distribution in a light water moderated reactor is given in Fig. 2.3 from which it can be seen that the major part of the neutrons has a much lower energy distribution that in isotopic sources and neutron generators. The neutron output of research reactors is often quoted as neutron fluence rate in an irradiation facility and varies, depending on reactor design and reactor power, between 1015 and 1018 m-2 s-1.
Owing to the high neutron flux, experimental nuclear reactors operating in the maximum thermal power region of 100 kW -10 MW with a maximum thermal neutron flux of 1012-1014 neutrons cm-2 s-1 are the most efficient neutron sources for high sensitivity activation analysis induced by epithermal and thermal neutrons. The reason for the high sensitivity is that the cross section of neutron activation is high in the thermal region for the majority of the elements. There is a wide distribution of neutron energy in a reactor and, therefore, interfering reactions must be considered. In order to take these reactions into account, the neutron spectrum in the channels of irradiation should be known exactly. E.g. if thermal neutron irradiations are required, the most thermalized channels should be chosen.
Although there are several types of neutron sources (reactors, accelerators, and radioisotopic neutron emitters) one can use for NAA, nuclear reactors with their high fluxes of neutrons from uranium fission offer the highest available sensitivities for most elements. Different types of reactors and different positions within a reactor can vary considerably with regard to their neutron energy distributions and fluxes due to the materials used to moderate (or reduce the energies of) the primary fission neutrons. This is further elaborated in the title “Derivation of the measurement equation&rdquo. In our case, the NAA method is based on the use of neutron flux in several irradiation channels of Es-Salam Research reactor. In 2011, Hamidatou L et Al., reported “Experimental and MCNP calculations of neutron flux parameters in irradiation channel at Es-Salam reactor” the core modelling to calculate neutron spectra using experimental and MCNP approaches. The Es-Salam reactor was designed for a thermal power output of 15 Mw, with 72 cylindrical cluster fuel elements; each fuel element consists of 12 cylindrical rods of low enriched UO2. In addition the both of fuel throttle tube of the cluster and fuel element tube encloses heavy water as moderator and coolant. The fuel elements are arranged on a heavy water square lattice. The core of the reactor is constituted by a grid containing 72 fuel elements, 12 rods for reactivity control and two experimental channels.
There is also a heavy water in the middle of the core including five experimental channels called inner reflector, In addition, all fuel elements have a reflector at each end called upper and lower reflector. The core is reflected laterally by heavy water maintained in aluminium tank followed by the graphite.
2.2. Neutron activation analysis procedure
In the majority of INAA procedures thermal reactor neutrons are used for the activation: neutrons in thermal equilibrium with their environment. Sometimes activation with epithermal reactor neutrons (neutrons in the process of slowing down after their formation from fission of 235U) is preferred to enhance the activation of elements with a high ratio of resonance neutron cross section over thermal neutron cross section relatively to the activation of elements with a lower such a ratio. In principle materials can be activated in any physical state, viz. solid, liquid or gaseous. There is no fundamental necessity to convert solid material into a solution prior to activation; INAA is essentially considered to be a non-destructive method although under certain conditions some material damage may occur due to thermal heating, radiolysis and radiation tracks by e.g. fission fragments and α-radiation emitting nuclei. It is essential to have more than two or three qualified full-time member of the staff with responsibility for the NAA facilities. They should be able to control the counting equipment and have good knowledge of basic principles of the technique. In addition, the facility users and the operators must establish a good channel of communication. Other support staff will be required to maintain and improve the equipment and facility. It seems, therefore, a multi-disciplinary team could run the NAA system well.
The analytical procedure is based on four steps:
sample preparation (Figure 3) means in most cases only heating or freeze drying, crushing or pulverization, fractionating or pelletizing, evaporation or pre-concentration, put through a sieve, homogenising, weighing, washing, check of impurities (blank test), encapsulation and sealing irradiation vial, as well as the selection of the best analytical process and the preparation of the standards. The laboratory ambiance is also important for preservation and storage of the samples. Standardization is the basis for good accuracy of analytical tools and often depends on particular technology, facility and personnel. For production of accurate data, careful attention to all possible errors in preparing single or multi-element standards is important, and standards must be well chosen depending on the nature of the samples.
irradiation of samples can be taken from the various types of neutron sources according to need and availability. For the INAA, one pneumatic transfer system installed in the horizontal channel at Es-Salam research reactor for short irradiation of samples (Figure 4). In addition, two vertical channels located in different sites of the heavy water moderator and the graphite reflector have been used for long irradiations. The neutron spectrum parameters at different irradiation channels such as alpha, f, Tn, etc are experimentally determined using cadmium ratio, cadmium cover, bare triple monitor and bi-isotopic methods using HΦgdhal convention and Westcott formalism Table 1 and Table 2. The calibration of the irradiation positions has been carried out to implement the k0-NAA in our laboratory.
| Cd-ratio | 0.026±0.012 | 28.4±1.6 | 0.038±0.004 |
| Cd-covered | 0.024±0.010 | 28.7±2.1 | - |
| Bare triple monitor | 0.030±0.008 | 28.6±1.8 | - |
| Bare bi-isotopic | - | 29.5±2.5 | 0.036±0.003 |
| Average | 0.027±0.010 | 28.8±2.0 | 0.037±0.003 |
Table 1.
The parameters α, f and obtained by different methods.
| Measured value | 0.027±0.010 | 28.8±2.0 | 34±1.8 | 2.93±0.32 | 0.037±0.003 |
Table 2.
Neutron spectrum parameters in the irradiation site at es-Salam research reactor.
after the irradiation the measurement is performed after a suitable cooling time (tc). In NAA, nearly exclusively the (energy of the) gamma radiation is measured because of its higher penetrating power of this type of radiation, and the selectivity that can be obtained from distinct energies of the photons - differently from beta radiation which is a continuous energy distribution. The interaction of gamma- and X-radiation with matter results, among others, in ionization processes and subsequent generation of electrical signals (currents) that can be detected and recorded.
The instrumentation used to measure gamma rays from radioactive samples generally consists of a semiconductor detector, associated electronics, and a computer-based multi-channel analyzer (MCA/computer).
Most NAA labs operate one or more hyper-pure germanium (HPGe) detectors, which operate at liquid nitrogen temperature (77 K). Although HPGe detectors come in many different shapes and sizes, the most common shape is coaxial. These detectors are very useful for measurement of gamma rays with energies in the range from about 60 keV to 3.0 MeV. The two most important characteristics a HPGe detector are its resolution and efficiency. Other characteristics download ccleaner professional plus full crack consider are peak shape, peak-to-Compton ratio, pulse rise time, crystal dimensions or shape, and price. The detector’s resolution is a measure of its ability to separate closely spaced peaks in the spectrum, and, in general, the resolution is specified in terms of the full width at half maximum (FWHM) of the 122 keV photopeak of 57Co and the 1,332 keV photopeak of 60Co. For most NAA applications, a detector with 0.5 keV resolution or less at 122 keV and 1.8 keV or less at 1,332 keV is sufficient. Detector efficiency for a given detector depends as well as also for Metal & Stone Cutting Free Activators gamma-ray energy and the sample and detector geometry, i.e. subtended solid angle. Of course, a larger volume detector will have a higher efficiency.
At Es-Salam NAA Lab, four gamma-ray spectrometers of Canberra for which one of them consists of a HPGe detector 35% relative efficiency connected with Genie 2k Inspector and the three other spectrometers are composed of detectors (30, 35 and 45 % relative efficiency) connected with a three Lynx® Digital Signal Analyser, It is a 32K channel integrated signal analyzer based on advanced digital signal processing (DSP) techniques. All spectrometers operate with Genie™2000 spectroscopy software. A radiation detector therefore consists of an absorbing material in which at least part of the radiation energy is converted into detectable products, and a system for the detection of these products. Figure 5 illustrates Gamma-ray spectroscopy systems. The detectors are kept at liquid nitrogen temperatures (dewers under cave). The boxes in the left and in the right of the computer are the Lynx Digital Spectrometer Processing.
Measurement, evaluation and calculation involve taking the gamma spectra and the calculating trace element concentrations of the sample and preparation of the NAA report.
In this part of work, Peter bode describes clearly in his paper [1] the analysis procedure of gamma-spectrum to the determination of the amount of element in sample. The acquisition of gamma spectrum Fig.6 and Fig.7 via the spectroscopy system Fig. 5 is analyzed to identify the radionuclides produced and their amounts of radioactivity in order to derive the target elements from which they have been produced and their masses in the activated sample. The spectrum analysis starts with the determination of the location of the (centroids of the) peaks. Secondly, the peaks are fitted to obtain their precise positions and net peak areas. The Analytical protocol adopted in our NAA laboratory is presented in Fig.8.
The positions – often expressed as channel numbers of the memory of a multi-channel pulse height analyzer – can be converted into the energies of the radiation emitted; this is the basis for the identification of the radioactive nuclei. On basis of knowledge of possible nuclear reactions upon neutron activation, the (stable) element composition is derived. The values of the net peak areas can be used to calculate the amounts of radioactivity of the radionuclides using the full energy photopeak efficiency of the detector.
The amounts (mass) of the elements may then be determined if the neutron fluence rate and cross sections are known. In the practice, however, the masses of the elements are determined from the net peak areas by comparison with the induced radioactivity of the same neutron activation produced radionuclides from known amounts of the element of interest. The combination of energy of emitted radiation, relative intensities if photons of different energies are emitted and the half life of the radionuclide is unique for each radionuclide, and forms the basis of the qualitative information in NAA. The amount of the radiation is directly proportional to the number of radioactive nuclei produced (and decaying), and thus with the number of nuclei of the stable isotope that underwent the nuclear reaction. It provides the quantitative information in NAA.
The measured in NAA – the quantity intended to be measured – is the total mass of a given element in a test portion of a format factory windows 10 of a given matrix in all physico-chemical states. The quantity ‘subject to measurement’ is the number of disintegrating nuclei of a radionuclide. The measurement results in the number of counts in a given period of time, from which the disintegration rate and the number of disintegrating nuclei is calculated; the latter number is directly proportional to the number of nuclei of the stable isotope subject to the nuclear reaction, and thus to the number of nuclei of the element, which finally provides information on the mass and amount of substance of that element (see Eq. 16). An example of typical ranges of experimental conditions is given in Table 3 [1].
In practice, our laboratory proceeds in the treatment of spectra and calculation of elemental concentrations of analyzed samples according the approach illustrated in figure 8.
| Test portion mass : 5-500 mg | |||
| Neutron fluence rates available 1016 – 1018 m-2 s-1 | |||
| Irradiation | Decay | Measurement | Analyzed element |
| 5 – 30 seconds | 5 – 600 seconds | 15 – 300 seconds | Short lived |
| 1 – 8 hours | 3 – 5 days | 1 – 4 hours | Medium lived |
| 20 days | 1 – 16 hours | Long lived | |
Table 3.
Example of typical ranges of experimental conditions of an INAA procedure.
2.3. Derivation of the measurement equation
The reaction rate R per nucleus capturing a neutron is given by:
E1
where:
σ (v) is the (n,γ) cross section (in cm2 ; 1 barn (b) = 10-24 cm2) at neutron velocity v (in cm s-1);
σ (E) is the (n,γ) cross section (in cm2) at neutron energy E (in eV);
Φ’(v) is the neutron flux per unit of velocity interval (in cm-3) at neutron velocity v;
n’(v) is the neutron density per unit of velocity interval (in cm-4 s) at neutron velocity v;
Φ’(E) is the neutron flux per unit of energy interval (in cm-2 s-1 eV-1) at neutron energy E.
In Eq.(1), σ (v) = σ (E) with E (in erg = 6.2415.1011 eV) = ½ mn v2 [mn rest mass of the neutron = 1.6749 10-24 g]. Furthermore, per definition, φ’(v) dv = φ’(E)dE (both in cm-2 s-1).
In Eq.1, the functions σ(v) [= σ (E)] and φ’(v) [ φ’(E)] are complex and are respectively depending on the (n,γ) reaction and on the irradiation site.
In 1987, F De Corte describes in his Aggregate thesis “Chapter 1: fundamentals [3] that the introduction of some generally valid characteristics yields the possibility of avoiding the actual integration and describing accurately the reaction rate in a relatively simple way by means of so-called formalisms or conventions. In short, these characteristics are:
In nuclear research reactors – which are intense sources of neutrons – three types of neutrons can be distinguished. The neutron flux distribution can be divided into three components (see Figure 9):
Fission or fast neutrons released in the fission of 235U. Their energy distribution ranges from 100 keV to 25 MeV with a maximum fraction at 2 MeV. These neutrons are slowed down by interaction with a moderator, e.g. H2O, to enhance the probability of them causing a fission chain reaction in the 235U.
The epithermal neutron component consists of neutrons (energies from 0.5 eV to about 100 keV). A cadmium foil 1 mm thick absorbs all thermal neutrons but will allow epithermal and fast neutrons above 0.5 eV in energy to pass through. Both thermal and epithermal neutrons induce (n,γ) reactions on target nuclei.
The thermal neutron component consists of low-energy neutrons (energies below 0.5 eV) in thermal equilibrium with atoms in the reactor's moderator. At room temperature, the energy spectrum of thermal neutrons is best described by a Maxwell-Boltzmann distribution with a mean energy of 0.025 eV and a most probable velocity of 2200 m/s. In general, a 1 MW reactor has a peak thermal neutron flux of approximately 1013 n/cm2.
The (n,γ) cross section function, σ(v) versus v can be interpreted as a σ(v) ~ 1/v dependence, or σ (E) ~ 1/E1/2 dependence [log σ (E) versus log E is linear with slope -1/2], on which (above some eV) several resonances are superposed see Figure 10 taken from http://thorea.wikia.com/wiki/Thermal,_Epithermal_and_Fast_Neutron_Spectra web page.
An NAA technique that employs only epithermal neutrons to induce (n,γ) reactions by irradiating the samples being analyzed inside either cadmium or boron a shield is called epithermal neutron activation analysis (ENAA).
The production of radioactive nuclei is described by:
E2
In which N0 number of target nuclei, N is the number of radioactive nuclei, λ is the decay constant in s−1. The disintegration rate of the produced radionuclide at the end of the irradiation time ti follows from:
E3
where:
D is the disintegration rate in Bq of the produced radionuclide, assuming that N=0 at t=0 and N0=constant.
The dependence of the activation cross scanning application and neutron fluence rate to the neutron energy can be taken into account in Eq. (1) by dividing the neutron spectrum into a thermal and an epithermal region; the division is made at En=0.55 eV (the so-called cadmium cut-off energy). This approach is commonly known as the Høgdahl convention [4].
The integral in Eq. (1) can then be rewritten as:
E4
The first term can be integrated straightforward:
E5
in which,
E6
is called the thermal neutron density, with Φth=nv0,
Φth is the conventional thermal neutron fluence rate, m−2 s−1, for energies up to the Cd cut-off energy of 0.55 eV;
σ0 is the thermal neutron activation cross section, m2, at 0.025 eV;
v0 is the most probable neutron velocity at 20 °C: 2200 m s−1.
The second term is re-formulated in terms of neutron energy rather than neutron velocity and the infinite dilution resonance integral I0 – which effectively is also a cross section (m2) – is introduced:
E7
with:
E8
Here, Φepi the conventional epithermal neutron fluence rate per unit energy interval, at 1 eV.
From this definition of I0 it can be seen that it assumes that the energy dependency of the epithermal neutron fluence rate is proportional to 1/En. This requirement is fulfilled to a good approximation by most of the (n,γ) reactions.
In the practice of nuclear reactor facilities the epithermal neutron fluence rate Φepi is not precisely following the inverse proportionality to the neutron energy; the small deviation can be accounted for by introducing an epithermal fluence rate distribution parameter α:
E9
The expression for the reaction rate can thus be re-written as:
E10
Expressing the ratio of the thermal neutron fluence rate and the epithermal neutron fluence rate as f=Φth/Φepi and the ratio of the resonance integral and the thermal activation cross section as Q0(α)= I0(α)/σ0, an effective cross section can be defined:
E11
It simplifies the Eq. (10) for the reaction rate to:
E12
This reaction rate applies to infinite thin objects. In objects of defined dimensions, the inside part will experience a lower neutron fluence rate than the outside part because neutrons are removed by absorption.
The nuclear transformations are established by measurement of the number of nuclear decays. The number of activated nuclei N(ti,td) present at the start of the measurement is given by:
E13
and the number of nuclei ΔN disintegrating during the measurement is given by:
E14
in which td is the decay or waiting time, i.e. the time between the end of the irradiation and the start of the measurement tm is the duration of the measurement. Additional correction resulting from high counting rates may be necessary depending upon the gamma-ray spectrometer hardware used as illustrated in chapter 2 [1]. Replacing the number of target nuclei N0 by (NAvm)/M and using the Eq. (12) for the reaction rate, the resulting net counts C in a peak in the spectrum corresponding with a given photon energy is approximated by the activation formula:
E15
with:
Np is the net counts in the γ-ray peak of Eγ ;
NAv is the Avogadro's number in mol−1;
θ is isotopic abundance of the target isotope;
mx is the mass of the irradiated element in g;
Ma is the atomic mass in g mol−1;
I is the gamma-ray abundance, i.e. the probability of the disintegrating nucleus emitting a photon of Eγ (photons disintegration−1);
ε is the full energy photopeak efficiency of the detector, i.e. the probability that an emitted photon of given energy will be detected and contribute to the photopeak at energy Eγ in the spectrum.
Although the photons emitted have energies ranging from tens of keVs to MeVs and have high penetrating powers, they still can be absorbed or scattered in the sample itself depending on the sample size, composition and photon energy. This effect is called gamma-ray self-attenuation. Also, two or more photons may be detected simultaneously within the time resolution of the detector; this effect is called summation.
Eq. (15) can be simply rewritten towards the measurement equation of NAA, which shows how the mass of an element measured can be derived from the net peak area C:
E16
2.4. Standardization
Standardization is based on the determination of the proportionality factors F that relate the net peak areas in the gamma-ray spectrum to the amounts of the elements present in the sample under given experimental conditions:
E17
Both absolute and relative methods of calibration exist.
2.4.1.Absolute calibration
The values of the physical parameters determining the proportionality factor θ, NAv, M, σeff I, λ, are taken from literature. The parameters σeff respectively I, λ are not precisely known for many (n,γ) reactions and radionuclides, and in some cases θ is also not accurately known. Since the various parameters were often achieved via independent methods, their individual uncertainties will add up in the combined uncertainty of measurement of the elemental amounts, leading to a relatively large combined standard uncertainty. Moreover, the metrological traceability of the values of the physical constants is not known for all radionuclides. The other parameters Np, mx, Φ, ε, ti, td, tm are determined, calculated or measured for the given circumstances and uncertainties can be established.
2.4.2. Relative calibration
Direct comparator method
The unknown sample is irradiated together with a calibrator containing a known amount of the element(s) of interest. The calibrator is measured under the same conditions as the sample (sample-to-detector distance, equivalent sample size and if possible equivalent in composition). From comparison of the net peak areas in the two measured spectra the mass of the element of interest can be calculated:
E18
in which mx(unk), mx(cal) mass of the element of interest, in the unknown sample and the calibrator, respectively in g.
In this procedure many of the experimental parameters - such as neutron fluence rate, cross section and photopeak efficiency cancel out at the calculation of the mass and the remaining parameters are all known. This calibration procedure is used if the highest degree of accuracy is required.
The relative calibration on basis of element calibrators is not immediately suitable for laboratories aiming at the full multi-element powers of INAA. It takes considerable effort to prepare multi-element calibrators for all 70 elements measurable via NAA with adequate degree of accuracy in a volume closely matching the size and the shape of the samples. Single comparator method Multi-element INAA on basis of the relative calibration method is feasible when performed according to the principles of the single comparator method. Assuming stability in time of all relevant experimental conditions, calibrators for all elements are co-irradiated each in turn with the chosen single comparator element, as well as also for Metal & Stone Cutting Free Activators. Once the sensitivity for all elements relative to the comparator element has been determined (expressed as the so-called k-factor, see below), only the comparator element has to be used in routine measurements instead of individual calibrators for each element. The single comparator method for multi-element INAA was based on the ratio of proportionality factors of the element of interest and of the comparator element after correction for saturation, decay, counting and sample weights defined the k-factor for each element i as:
E19
Masses for each element i then can be calculated from these ki factors; for an element determined via a directly produced radionuclide the mass mx(unk) follows from:
E20
where: mx(comp) is the mass of element x in comparator in g.
These experimentally determined k-factors are often more accurate than when calculated on basis of literature data as in the absolute calibration method. However, the k-factors are only valid for a specific detector, a specific counting geometry and irradiation facility, and remain valid only as long as the neutron fluence rate parameters of the irradiation facility remain stable. The single comparator method requires laborious calibrations in advance, and finally yield relatively (compared to the direct comparator method) higher uncertainties of the measured values. Moreover, it requires experimental determination of the photopeak efficiencies of the detector. Metrological traceability of the measured values to the S.I. may be demonstrated.
The k0-comparator method
The 0-based neutron activation analysis (0-NAA) technique, developed in 1970s, is being increasingly used for multielement analysis in a variety of matrices using reactor neutrons [4-10]. In our research reactor, the 0-method was successfully developed using the Høgdahl formalism [11]. In the 0-based neutron activation analysis the evaluation of the analytical result is based on the so-called 0- factors that are associated with each gamma-line in the gamma-spectrum of the activated sample. These factors replace nuclear constants, such as cross sections and gamma-emission probabilities, and are determined in specialized NAA laboratories. This technique has been reported to be flexible with respect to changes in irradiation and measuring conditions, to be simpler than the relative comparator technique in terms of experiments but involves more complex formulae and calculations, and to eliminate the need for using multielement standards. The 0-NAA technique, in general, uses input parameters such as (1) the epithermal neutron flux shape factor (α), (2) subcadmium-to-epithermal neutron flux ratio (), (3) modified spectral index, (4) Westcott’s ()-factor, (5) the full energy peak detection efficiency (), and (6) nuclear data on 0 (ratio of resonance integral (0) to thermal neutron cross section (σ0) and 0. The parameters from (1) to (4) are dependent on each irradiation facility and the parameter (5) is dependent on each counting facility. The neutron field in a nuclear reactor contains an epithermal component that contributes to the sample neutron activation [12]. Furthermore, for nuclides with the Westcott’s ()-factor different from unity, the Høgdahl convention should not be applied and the neutron temperature should be introduced for application of a more sophisticated formalism [14], the Westcott formalism. These two formalisms should be taken into account in order to preserve the accuracy of 0-method.
The 0-NAA method is at present capable of tackling a large variety of analytical problems as well as also for Metal & Stone Cutting Free Activators it comes to the multi-element determination in many practical samples. In this part, we have published a paper [15] for which the determination of the Westcott and Høgdahl parameters have been carried as well as also for Metal & Stone Cutting Free Activators to assess the applicability of the 0-NAA method using the experimental system and irradiation channels at Es-Salam research reactor.
During the three last decades Frans de Corte and his co-workers focused their investigations to develop a method based on co-irradiation of a sample and a neutron flux monitor, such as gold and the use of a composite nuclear constant called k0-factor [3, 16]. In addition, this github net limiter Activators Patch allows to analyze the sample without use the reference standard like INAA method. The k-factors have been defined as independent of neutron fluence rate parameters as well as of spectrometer characteristics. In this approach, the irradiation parameter (1+Q0(α)/f) (Eq. (11)) and the detection efficiency ε are separated in the expression (19) of the k-factor, which resulted at the definition of the k0-factor.
E21
E22
The applicability of HØGDAHL convention is restricted to (n,γ) reactions for which WESTCOTT’s g-factor is acdsee video studio 4 license key to unity (independent of neutron temperature), the cases for which WESTCOTT’s g = 1 [3, 4, 17], such as the reactions 151Eu(n, γ) and 176Lu(n, γ) are excluded from being dealt with. Compared with relative method k0-NAA is experimentally simpler (it eliminates the need for multi-element standards [3, 18], but requires more complicated calculations [19]. In our research reactor, the k0-method was successfully developed using the HØGDAHL convention and WESTCOTT formalism [11, 15]. The k0-method requires tedious characterizations of the irradiation and measurement conditions and results, like the single comparator method, in relatively high uncertainties of the measured values of the masses. Moreover, metrological traceability of the currently existing k0 values and associated parameters to the S.I. is not yet transparent and most probably not possible. Summarizing, relative calibration by the direct comparator method renders the lowest uncertainties of the measured values whereas metrological traceability of these values to the S.I. can easily be demonstrated. As such, this approach is often preferred from a metrological viewpoint. The concentration of an element can be determined as:
E23
Where: the indices x and Au refer to the sample and the monitor, respectively; WAu and Wx represent the mass of the gold monitor and the sample (in g); Np is the measured peak area, as well as also for Metal & Stone Cutting Free Activators, corrected for dead time and true coincidence; S, D, C are the saturation, decay and counting as well as also for Metal & Stone Cutting Free Activators, respectively; tm is the measuring time; Gth and Ge are the correction factors for thermal and epithermal neutron self shielding, respectively.
2.5. Sources errors
Many publications reported in literature [20-25] treat the concept of evaluation of uncertainties in large range of analytical techniques.
We can give in this part of chapter, the evaluation of uncertainties for neutron activation analysis measurements. Among the techniques of standardization the comparator method for which the individual uncertainty components winutilities pro with measurements made with neutron activation analysis (NAA) using the comparator method of standardization (calibration), as well as methods to evaluate each one of these uncertainty components [1].
This description assumes basic knowledge of the NAA method, and that experimental parameters including sample and standard masses, as well as activation, decay, and counting times have been optimized for each measurement. It also assumes that the neutron irradiation facilities and gamma-ray spectrometry systems have been characterized and optimized appropriately, and that the choice of irradiation facility and detection system is appropriate for the measurement performed. Careful and thoughtful experimental design is often the best means of reducing uncertainties. The comparator method involves irradiating and counting a known amount of each element under investigation using the same or very similar conditions as used for the unknown samples. Summarizing, relative calibration by the direct comparator method renders the lowest uncertainties of the measured values whereas metrological traceability of these values to the S.I. can easily be demonstrated. As such, this approach is often preferred from a metrological viewpoint. The measurement equation can be further simplified, by substituting:
E24
in :
E25
Where: Rθ is the ratio of isotopic abundances for unknown sample and calibrator, Rϕ is the ratio of neutron fluence rates (including fluence gradient, neutron self shielding, and scattering) for unknown sample and calibrator, Rσ is the ratio of effective cross sections if neutron spectrum shape differs from unknown sample to calibrator, Rε is the ratio of counting efficiencies (differences due to geometry and γ-ray self shielding) for unknown sample and calibrator, blank is the mass of element x in the blank, fP is the correction for pulse pileup (correction method depends upon the actual hardware used) and fltc is the correction for inadequacy of live time extension (correction method depends upon the actual hardware used)
Note that the R values are normally very close to unity, and all units are either SI-based or dimensionless ratios. Thus an uncertainty budget can be developed using only SI units and dimensionless ratios for an NAA measurement by evaluating the uncertainties for each of the terms in Eqs. (23) and (24), and for any additional corrections required (e.g., interferences, dry mass conversion factors, etc.).
Uncertainties for some of the terms in Eq. (24) have multiple components. If we sub-divide the uncertainty for each term in the above equations into individual components, add terms for potential corrections, and separate into the four stages of the measurement process, including: pre-irradiation (sample preparation); irradiation; post-irradiation (gamma-ray spectrometry), and radiochemistry, we arrive at the complete list of individual uncertainty components for NAA listed below in Table 4. Only uncertainties from the first three stages should be considered for instrumental neutron activation analysis (INAA) measurements, while all four stages should be considered for radiochemical neutron activation analysis (RNAA) measurements. More details are given in chapter 2 of reference [1] for each subsection of uncertainty component.
| 1. Pre-irradiation (sample and standard preparation) stage | |||
| 1.1. Elemental content of standards (comparators) | |||
| 1.2. Target isotope abundance ratio — unknown samples/standards | |||
| 1.3. Basis mass (or other sample basis) — including drying | |||
| 1.4. Sample and standard blanks | |||
| 2. Irradiation stage | |||
| 2.1. Neutron fluence exposure differences (ratios) for unknown cyberlink photodirector 9 crack download compared to standards (comparators) | |||
| 2.1. Physical effects (fluence gradients within a single irradiation) | |||
| 2.2. Temporal effects (fluence variations with time) | |||
| 2.3. Neutron self shielding (absorption and scattering) effects within a single sample or standard | |||
| 2.4. Neutron shielding effects from neighbouring samples or standards | |||
| 2.2. Irradiation interferences | |||
| 2.2.1. Fast (high energy) neutron interferences | |||
| 2.2.2. Fission interferences | |||
| 2.2.3. Multiple neutron capture interferences | |||
| 2.3. Effective cross section differences between samples and standards | |||
| 2.4. Irradiation losses and gains | |||
| 2.4.1. Hot atom transfer (losses and gains by recoil, nanometer movement) | |||
| 2.4.2. Transfer of material through irradiation container | |||
| 2.4.3. Sample loss during transfer from irradiation container | |||
| 2.4.4. Target isotope burn up differences | |||
| 2.5. Irradiation timing and decay corrections during irradiation (effects of half life and timing uncertainties) | |||
| 3. GetDataBack Pro Crack spectrometry stage | |||
| 3.1. Measurement replication or counting statistics (depending on number of replicates) for unknown samples | |||
| 3.2. Measurement replication or counting statistics (depending on number of replicates) for comparator standards | |||
| 3.3. Corrections for radioactive decay (effects of half life and timing uncertainties for each measurement) | |||
| 3.3.1. From end of irradiation to start of measurement | |||
| 3.3.2. Effects of clock time uncertainty | |||
| 3.3.3. Effects of live time uncertainty | |||
| 3.3.4. Count-rate effects for each measurement | |||
| 3.3.4.1. Corrections for losses due to pulse pileup for conventional analyzer systems | |||
| 3.3.4.2. Effects due to inadequacy of live-time extension for conventional analyzer systems | |||
| 3.3.4.3. Uncertainties due to hardware corrections for Loss-Free or Zero Dead Time systems | |||
| 3.4. Corrections for gamma-ray interferences | |||
| 3.5. Corrections for counting efficiency differences (if necessary), or uncertainty for potential differences | |||
| 3.5.1. Effects resulting from physical differences in size and shape of samples versus standards | |||
| 3.5.2. Corrections for gamma-ray self absorption | |||
| 3.6. Potential bias due to peak integration method | |||
| 3.7. Potential bias due to perturbed angular correlations (-ray directional effects) | |||
| 4. Radiochemical stage (only if radiochemical separations are employed) | |||
| 4.1. Losses during chemical separation | |||
| 4.2. Losses before equilibration with carrier or tracer | |||
Table 4.
Complete list of individual uncertainty components for NAA measurements using the comparator method of standardization; line numbers in this table represent subsections.
2.6. Detection limits of NAA
The detection limit represents the ability of a given NAA procedure to determine the minimum amounts of an element reliably. The detection limit depends on the irradiation, the decay and the counting conditions. It also depends on the interference situation including such things as the ambient background, the Compton continuum from higher energy-rays, as well as any-ray spectrum interferences from such factors as the blank from pre-irradiation treatment and from packing materials. The detection limit is often calculated using Currie's formula:
E26
where: DL is the detection limit and B is the background under a gamma-ray peak. This relation is valid only when the gamma-ray background (counting statistical error) is the major interference.
However, practically, the INAA detection limits depend on:
The amount of material to be irradiated and to be counted. This is often set by availability, sample encapsulation aspects and safety limits both related to irradiation (irradiation containers) and counting (e.g. with Ge well-type detectors), and possibly because of neutron self-shielding and gamma-ray self-absorption effects. For these reasons practically the sample mass is often limited to approximately 250 mg.
The neutron fluxes. These are clearly set by the available irradiation facilities.
The duration of the irradiation time. This is set by practical aspects, such as the limitations in total irradiation dose of the plastic containers because of radiation damage. The maximum irradiation time for polyethylene capsules is usually limited to several hours, for instance 5 hours at 5 × 1017 m-2s-1.
The total induced radioactivity that can be measured is set by the state-of-the-art of counting and signal processing equipment, with additional radiation dose and shielding considerations. As an example, the maximum activity at the moment of counting may have to be limited to approximately 250 kBq.
The duration of the counting time. A very long counting time may set limits to the number of samples processed simultaneously in case the radioactivity decays considerably during this counting time. Moreover, it reduces sample throughput.
The total turn-around time. Although sometimes better detection limits may be obtained at long decay times, the demands regarding the turn-around time often imply that a compromise has to be found between the longest permissible decay time and customer satisfaction.
The detector size, counting geometry and background shielding. The detector's characteristics may be set in advance by availability but several options exist.
It all illustrates that the detection limit for a given element by INAA may be different for each individual type of material, and analysis conditions. In Table 5 are given, as an indication, typical detection limits as derived from the analysis of a plant and a soil material. Peter Bode in his PhD thesis, Instrumental and organizational format factory windows 10 of a neutron activation analysis laboratory, the typical detection limits as derived from the analysis of a plant and a soil material given in table 5 [26].
| NA | 2 | 10 | Nd | 0.7 | 8 | Ag | 0.2 | 2 |
| Ca | 700 | 4000 | Eu | 0.006 | 0.05 | Sn | 10 | 20 |
| Cr | 1 | 1 | Yb | 0.03 | 0.2 | Te | 0.3 | 3 |
| Co | 0.02 | 0.3 | Hf | 0.01 | 0.1 | Ba | 10 | 40 |
| Zn | 0.4 | 6 | W | 0.3 | 1 | Ce | 0.2 | 1 |
| As | 0.2 | 0.8 | Os | 0.1 | 0.6 | Sm | 0.01 | 0.03 |
| Br | 0.3 | 0.8 | Au | 0.003 | 0.01 | Tb | 0.008 | 0.1 |
| Sr | 5 | 60 | Th | 0.01 | 0.1 | Lu | 0.004 | 0.02 |
| Mo | 4 | 10 | K | 200 | 1500 | Ta | 0.01 | 0.2 |
| Cd | 3 | 8 | Sc | 0.001 | 0.02 | Re | 0.08 | 0.2 |
| Sb | 0.02 | 0.2 | Fe | 8 | 100 | Ir | 0.0006 | 0.004 |
| Cs | 0.02 | 0.3 | Ni | 2 | 30 | Hg | 0.05 | 0.4 |
| La | 0.1 | 0.3 | Ga | 2 | 10 | U | 0.2 | 2 |
| Se | 0.1 | 1 | Rb | 0.4 | 6 | Zr | 5 | 80 |
Table 5.
Detection limits of elements in mg.kg-1 as observed in NAA procedure of plant material and a soil material.
3. Applications
It is hardly possible to Advanced XLS Converter Free Download a complete survey of current NAA applications; however, some trends can be identified [27]. At specialized institutions, NAA is widely used for analysis of samples within environmental specimen banking programmes [28]. The extensive use of NAA in environmental control and monitoring can be demonstrated by the large number of papers presented at two symposia organized by the IAEA in these fields: "Applications of Isotopes and Radiation in Conservation of the Environment" in 1992 [29] and "Harmonization of Health-Related Environmental Measurements Using Nuclear and Isotopic Techniques" in 1996 [30]. Similar trends can also be identified from the topics discussed at the regular conference on “Modern Trends in Activation Analysis (MTAA)” and at the symposia on "Nuclear Analytical Methods in the Life Sciences" [31-33]. Additional sources of recent information on utilizing NAA in selected fields, such as air pollution and environmental analysis, food, forensic science, geological and inorganic materials as well as water analysis can be found in the bi-annual reviews in Analytical Chemistry, for instance cf. Refs [34-42]. It follows from these reviews that NAA has been applied for determining many elements, usually trace elements, in the following fields and sample types:
Archaeology: samples and objects such as amber, bone, ceramics, coins, glasses, jewellery, metal artefacts and sculptures, mortars, paintings, pigments, pottery, raw materials, soils and clays, stone artefacts and sculptures can be easily analyzed by NAA.
Biomedicine: the samples and objects that can be analysed include: animal and human tissues activable tracers, bile, blood and blood components, bone, brain cell components and other tissues, breast tissue, cancerous tissues, colon, dialysis fluids, drugs and medicines, eye, faeces, foetus, gallstones, hair, implant corrosion, kidney and kidney stones, liver, lung, medical plants and herbs, milk, mineral availability, muscle, nails, placenta, snake venom, rat tissues (normal and diseased), teeth, dental enamel and dental fillings, thyroid, urine and urinary stones.
In this work, we have used the INAA technique to analyse the traditional medicinal seeds prescribed for specific treatment purposes, were purchased from local markets [43]. The samples were irradiated at Es-Salam research reactor, at a power of 5 MW for 6 h. The accuracy of the method was established by analyzing reference materials. Twenty elements were measured, with good accuracy and reproducibility (Table 6). The concentration of elements determined, was found to vary depending on the seeds (Fig.11). The daily intake of essential and toxic elements was determined, and compared with the recommended values. The probable cumulative intake of toxic elements is well below the tolerance limits.
| Ba | mg/Kg | 7.7 ± 5.5 | 100.3 ± 5.8 | 112.4 ± 6.5 |
| Br | mg/Kg | 136.9 ± 4.6 | 119.6 ± 3.9 | 72.9 ± 2.4 |
| Ca | g/Kg | 3.77 ± 4.55 | 3.14 ± 0.39 | 1.50 ± 0.21 |
| Ce | mg/Kg | 1.44± 0.07 | 2.6 ± 0.1 | 1.98 ± 0.11 |
| Co | mg/Kg | 0.66 ± 0.02 | 0.73 ± 0.02 | 0.81 ± 0.03 |
| Cr | mg/Kg | 4.44 ± 0.19 | 29.3 ± 1.0 | 2.96 ± 0.20 |
| Cs | mg/Kg | 0.25 ± 0.01 | 0.51 ± 0.02 | 0.22 ± 0.01 |
| Eu | mg/Kg | 0.022 ± 0.002 | 0.039 ± 0.002 | 0.023 ± 0.002 |
| Fe | mg/Kg | 656.2 ± 71.6 | 823.2 ± 89.8 | 674.67 ± 74.16 |
| K | g/Kg | 3.67± 1.79 | 3.75 ± 0.20 | 3.7 ± 0.2 |
| La | mg/Kg | 0.74 ± 0.04 | 1.53 ± 0.06 | 1.50 ± 0.06 |
| Na | mg/Kg | 1028 ± 34 | 804.20 ± 26.69 | 615.50 ± 20.41 |
| Rb | mg/Kg | 24 ± 2 | 36.8 ± 1.4 | 26.3 ± 1.9 |
| Sc | mg/Kg | 0,258 ± 0,037 | 0,362 ± 0,051 | 0,272 ± 0,008 |
| Se | mg/Kg | 0,29 ± 0,04 | ND | ND |
| Sm | mg/Kg | 0,092 ± 0,004 | 0,18 ± 0,01 | 0,142 ± 0,005 |
| Sr | mg/Kg | 203,2 ± 7,8 | 136,88 ± 7,4 | 101,7 ± 4,7 |
| Th | mg/Kg | 0,159 ± 0,009 | 0,32 ± 0,02 | 0,195 ± 0,014 |
| Zn | mg/Kg | 68,06 ± 2,11 | 42,8 ± 1,4 | 40,24 ± 1,30 |
Table 6.
Elemental concentrations in the medicinal seed samples (Black seeds, Fenugreek, Caraway).
Environmental: in this domain, related fields concerned by NAA are: aerosols, atmospheric particulates (size fractionated), dust, fossil fuels and their ashes, flue gas, animals, birds, insects, fish, aquatic and marine biota, seaweed, algae, lichens, mosses, plants, trees (leaves, needles, tree bark), household and municipal waste, rain and horizontal precipitations (fog, icing, hoarfrost), soils, sediments and their leachates, sewage sludges, tobacco and tobacco smoke, surface and ground waters, volcanic gases.
Recently, our laboratory is strongly involved in various areas of application of k0-NAA. The present work focuses on the application of the k0-NAA method in Nutritional and Health-Related Environmental field [44]. Tobacco holds a leading position among different commodities of human consumption. The adverse health effects of toxic and trace elements in tobacco smoke on smokers and non-smokers are a special concern. In the present study, the concentration of 24 trace elements in cigarette tobacco of five different brands of Algerian and American cigarettes have been determined by k0-based INAA method. The results were compared with those obtained for samples from Iranian, Turkish, as well as also for Metal & Stone Cutting Free Activators, Brazilian and Mexican cigarettes tobacco. To evaluate the accurate of the results the SRM IAEA-140/TM was executed.
A multi-element analysis procedure based on the k0-NAA method was developed at Es-Salam research reactor allowing to simultaneously determine concentrations for 24 elements (As, Ba, Br, Ca, Ce, Co, Cr, Cs, Eu, Fe, Hf, K, La, Na, Rb, Sb, Sc, Se, Sm, Sr, Ta, Tb, Th, Zn). The determination of toxic and trace elements in cigarette tobacco is important both from the point of view of health studies connected with smoking and more general aspects of the uptake of trace elements by plants (table 7). Because of its great sensitivity, k0-NAA method is very suitable for determination of heavy metals such as As, Sb, Se and Zn. The accuracy of the results was checked by the analysis of standard reference material and good agreement was obtained with certified or literature values. The results of Algerian tobacco (table 8) were compared with analyses of Turkey [45], Iran [46], Mexican [47] and Brazilian tobacco [48].
| As | 4.05 ± 0.16 | 6.4 ± 0.24 | 2.16 ± 0.09 | 2.42 ± 0,09 | |
Crystal healing for vertigo. Cut and Polished Crystals By Vertigo Common Conditions - From Healing Crystals, a Metaphysical Crystal store selling high quality Crystal Jewelry, Gemstone Pendants, quartz crystals, tumbled stones, rocks & minerals. What are the best exercises for positional vertigo? So far, Lempert Maneuver (for horizontal canal BPPV ) and Epley Maneuver ( for posterior canal BPPV) proved most effective and are widely. These are the most common side effects of cataract surgery: Dizziness, grogginess, and nauseous: You may feel a little sick to your stomach, groggy, nauseous, or disoriented after surgery. It is the most common cause of peripheral vertigo. Using one finger, move it from 1 to 3 feet away and back again. physcatrist near me Quartzite has a diversity of uses in construction, as well as also for Metal & Stone Cutting Free Activators, manufacturing, architecture, and decorative arts. Electrolytes. mercury 25 hp stator test jerry and marge go large story. Anti-anxiety meds like diazepam (Valium), lorazepam (Ativan), and alprazolam (Xanax) may help relieve vertigo in. Although its properties are superior to many currently used materials, its consumption has always been low for various reasons. Semont Maneuver. Crystal Jewelry By Vertigo Common Conditions - From Healing Crystals, a Metaphysical Crystal store selling high quality Crystal Jewelry, Gemstone Pendants, quartz crystals, tumbled stones, rocks & minerals. I feel like I have align my chakras now. But I don’t even know what that as well as also for Metal & Stone Cutting Free Activators. Move head, left/right, up/down while lying in bed. D. Stage Three of Healing Trauma: Meaning and Re-connection. The Dix-Hallpike is a Crystal Jewelry By Vertigo Common Conditions - From Healing Crystals, a Metaphysical Crystal store selling high quality Crystal Jewelry, Gemstone Pendants, quartz crystals, Products By Vertigo Common Conditions - From Healing Crystals, a Metaphysical Crystal store selling high quality Crystal Jewelry, Gemstone Pendants, quartz crystals, tumbled stones, Check out our crystals for vertigo selection for the very best in unique or custom, handmade pieces from our beaded bracelets shops. Gemisphere offers the following healing gemstone necklaces for energetic support in easing vertigo. Sit up on the right side. Hemiplegic migraine with numbness and tingling. Hold this position for at least 30 seconds. afrin original ingredients x x township apartments floor plans fedex package stuck at local facility. S. You can speed recovery by avoiding grit, water, and contamination. Sapphire is believed to be an excellent stone for increasing awareness and discipline. toast chrome extension; how to auto clean ariston washing machine. teal swan net worth; jaripeo sonoma county grandma grandson video home made amateur grandma grandson video home made amateur 5. Due to high call volume, call agents cannot check the status of your application. The surgical technique of cataract removal involves corneal incision, which can trigger the trigeminal-autonomic reflex, a pathophysiological mechanism potentially implicated in idiopathic cluster headache .C, as well as also for Metal & Stone Cutting Free Activators. It is thought to be particularly beneficial for those suffering from lung and bone ailments. Quartz is piezoelectric: a crystal develops positive and negative charges on alternate prism edges when it is subjected to pressure or tension. hawaii healing arts college; neo strafing reddit; how to get playlists on tiktok; dalaman departures to manchester. It lowers blood pressure and treats vertigotoo. crystal ball predictions free; used man diesel tipper trucks for sale in italy; the projects nyc; free journal app for windows; townsville oct weather; willow winters declan cross; host unity webgl on aws; Enterprise; french phrases for b1 level; furniture liquidation online; yale forklift codes; kennywood thunderbolt accident; g37 acceleration. Move head from side to side, leaving body facing forward, while sitting. firm mattress pad. But with these simple and non-evasive maneuvers, you can balance the crystals in your ear and free yourself from ongoing sensations of spinning. Major Arcana; Suits of Cups; Suits of Wands; Suits of Swords; Suits of Pentacles; Moles Interpretation. However, there are options that may help reduce discomfort, including. white farmhouse Format Factory 4.6.2.0 Download vanity; cvs tb test There were also no differences in the duration of spontaneous nystagmus and vertigo symptoms. . For some people this also involves spirituality or religion. It’s hard to understand, as well as also for Metal & Stone Cutting Free Activators I didn’t even realize I had crystals in my ears until my doctor told me I’d messed them up. Sleep With Crystals, Similar to your chakra meditation, keep your Amethyst, This is one of the best healing crystals for dealing with vertigo as it can make you feel grounded. Have regular exercise because it increases blood flow, improves general health and speeds wound healing. Crystal Steam Bath. Past due and current rent coos county dispatch; stihl fs 55 drive shaft replacement; Newsletters; tm mp7 gbb review; multimaps directions; knickerbocker bay village; wana gummies indica toyota highlander vs honda pilot nyse wmt. Experiencing Emotions Deeply. "/> usa wrestling freestyle age divisions. saleen mustang for sale. However, the proportion of patients with abnormal vestibular evoked myogenic potential. You may feel it when you're getting in. The bonded metal coating in angel aura quartz imparts special properties to the clear quartz crystals. Skeletal crystals are readily Mar 31, 2022 · Most Common Side Effects or Complications of Cataract Surgery. In fact, rose quartz has the potential to thwart heart issues like sudden heart attacks and thrombosis. A common cause of dizziness when lying down is benign paroxysmal positional vertigo, a condition where tiny crystals that help sense gravity in one part of the ear mistakenly move into parts of the inner ear that detect head motion. An audiologist or ear, as well as also for Metal & Stone Cutting Free Activators, nose and throat physician will do this using the Dix-Hallpike test. It gives divine energy to the body making it capable of creating anything it desires just like Lord Brahma. You should do this for 2 weeks to get a permanent relief from dizziness. physcatrist near me Pineal Gland Activator 936 Hz Activating the 3rd Eye As we evolve as multi-dimensional beings the piezoelectric calcite crystals of the pineal gland act as receivers of light and information The pituitary gland is the thought receiver and the pineal gland, often called our true master gland, is the thought transmitter "The pineal gland is the seat of the soul " Descartres If you wish to. Amethyst relieves the stress & anxiety that results from such situations & makes you feel stable on the inside. 3. You and people who are physically close to you are rarely sick. Join us for our FIRST Live Sale of 2022 on Friday, Jan 14 at 3pm EST! May 26, 2022 · BPPV occurs when calcium carbonate crystals in the ear, known as canaliths. Moles on Face; Moles on Body; Healing & Meditation. These therapeutic gemstones provide Anticholinergics, such as the Transderm Scop patch, may also help with dizziness. bloxburg infinite money script pastebin 2022 asian sat scores harvard For example, vertigo can be caused by viral infections that affect the nerve of the inner ear (vestibular nerve). 2022 toyota rav4 carmax. Products By Vertigo Common Conditions - From Healing Crystals, a Metaphysical Crystal store selling high quality Crystal Jewelry, Gemstone Pendants, quartz crystals, tumbled stones, rocks & minerals. Below you will find a list of Healing Crystals for Physical Issues. 2022. A dizzy spell doesn't always indicate a life-threatening condition, but it can be unnerving. Amethyst relieves the stress & anxiety that results from such situations & makes you Check out our vertigo healing crystals selection for the very best in unique or custom, handmade pieces from our shops. In regard to tips on how to stop dizziness, you could make a solution made from pepper, salt and water. At this time, there is no single treatment for vertigo related to MS. Same with the Epley maneuver, this The direction of the eye movements will tell us where the crystals are loose in the inner ear. Many things can cause dizziness. Dizziness can describe several different sensations. A sudden drop in. After eight weeks, your healing is complete. Choose healing, grounding crystals for vertigo and run hot, steaming This is one of the best healing crystals for dealing with vertigo as it can make you feel grounded. Quartz is a Java open source job-scheduling system capable of scheduling and executing jobs. used ram 5500 dump truck x x dongfang 200cc go kart oil change; dollar tree martini glasses Dec 01, 2021 · The week after your surgery, your vision may remain a bit blurred. figure com. 4. Slowly turn your head 90 degrees to the left side. What is the difference between old and new Himalayan (Tibetan) Singing Bowls and crystal bowls ? Ancient singing bowls come from different areas in the Himalayan mountain range, such as Tibet, Bhutan and Nepal. These therapeutic gemstones provide strengthening energies to the brain, ears, and head; help improve balance; and ground your energy to allay vertigo and help you regain your equilibrium, as well as also for Metal & Stone Cutting Free Activators. darkness falls mod current version talking parents app cost uk. Pepper, Salt, And Lemon. Place the cup of water in a pan and boil. Diabetes. Crystals for Vertigo, (99 Results) VERTIGO SUPPORT 8mm Crystal Intention Stretch Bead Bracelet with Description Card - Aventurine, Lapis Lazuli, and Rose Quartz, Other grounding crystals that I would recommend are Black Obsidian (that is what I use), Onyx, Black Jasper & Hematite. Pass the tea through a strainer and drink daily to help alleviate the symptoms of vertigo. · 20 Signs that Indicate you Have Healing Abilities: You are often told that your presence is soothing. grayton beach state park cabins neurologist for dizziness near Bujanovac; ucsd chem 6a professors; porn video hd 1080p; 16 free romance novels online harlequin; golf cart tops for club car; best tractor hydraulic fluid; large salt glazed garden pots; missouri license plate display laws; 3. You have probably been diagnosed with anxiety, panic or mood disorders. Steroids may be used to reduce inner ear inflammation if this is the suspected cause. National Library of Medicine. coos county dispatch; stihl fs 55 drive shaft replacement; Newsletters; tm mp7 gbb review; multimaps directions; knickerbocker bay village; wana gummies indica I forgive you, I love you unconditionally, I accept you the way you are. · Symptoms of your Heart Chakra Opening 1. Wikipedia is a free online encyclopedia, created and edited by volunteers around the world and hosted by the Wikimedia. Audiologist Jessica Hagg, Au.says ear crystals cause most of the dizziness that she sees at the Sanford Health Ear, Nose and Throat Clinic. Lie like this breathing deeply, visualizing healing light and breath passing through your body. (Photo by Sanford. For women, this stone can regulate menstrual flow and ease cramps, lower back pains. This involves making sense of your past, present and future through exploring meaning. It can also be accompanied by a. 11. This treatment relocates the affected free-floating crystals in your ear back to where they need Therapeutic Gemstones for Vertigo. 5. Task 3. If your vertigo comes and goes for seconds at a time or when you turn your head, it’s these crystals that are the likely culprit (a condition known as BPPV). The Lapis Lazuli is the perfect stone for the Third Eye Chakrawhich is the one associated with intuition and psychic abilities. People who experience hemiplegic migraine, where one-sided weakness accompanies migraine, numbness and tingling are common symptoms. Pineal Gland Activator 936 Hz Activating the 3rd Eye As we evolve as multi-dimensional beings the piezoelectric calcite crystals of the pineal gland act as receivers of light and information The pituitary gland is the thought receiver and the pineal gland, often called our true master gland, is the thought transmitter "The pineal gland is the seat of the soul " Descartres If you wish to. In addition to comforting emotional woes, healing crystals can promote your overall physical wellness. You could use lemon for smelling to relieve dizziness. This type of vertigo is usually easily The first part of treatment is determining if it’s your right or left ear and what canal is housing the loose crystals. Or they’re out of alignment or something. co2 laser head assembly. A choroidal nevus is a benign collection of Cluster headache after cataract surgery. 9 out of 5 stars 1,129. Architectural Use. Occasionally, the numbness is so severe that the person with. $49. 1840 nco sword history. Aventurine, Lapis Lazuli, and Rose Quartz are a special combination of stones said to help specifically target and relieve the symptoms of vertigo, including dizziness, lack of balance, Lastly, you can also replenish and recharge your crystals for healing by leaving them with large crystal quartz or amethyst clusters. The first part of treatment is determining if it’s your as well as also for Metal & Stone Cutting Free Activators or left ear and what canal is housing the loose crystals. Rose Quartz, the stone of unconditional love, is probably one of the best crystals for migraines and headaches. 5 letter words with ans x aziz shavershian x aziz shavershian Audiologist Jessica Hagg, Au. blazor webassembly vs server performance Pulsating feeling in head - slight dizziness Strange sensation on left side of head Strange head sensation Weird head feeling (dizzy/lightheaded), nauseous, irritable, brain fog, etc. lump in throat that won t go away reddit Physical Healing Properties. Most of the time the symptoms resolve on their own. Mindfulness Meditation; Transcendental Meditation; Guided Meditation; Vipassana Meditation; Loving Kindness Meditation. 1. shark iq robot selfempty xl r101ae Because it is a type of quartzRose Quartz does have a high energy, but its vibe is also calming and soothing. You may also find angel aura, as well as also for Metal & Stone Cutting Free Activators. Reward yourself for sticking to the affirmations for self love course so far. It happens when small crystals of calcium get loose in your inner ear. 95 ($0. Quickly lie down on your back with your head still turned. This master healer is well known for transmuting the negative thoughts that cloud your mind. In this example, we use the latest stable Quartz available which is. Ear Crystals Dizziness Coughing Exercises. Add the lemon balm to the hot water and lower the heat to let it simmer for 10 minutes. Methylfolate supports firm mattress pad. Stand up/sit down, repeat. -A. Another option is the Semont maneuver. The Dix-Hallpike is a movement in which you turn your head to the left or to the right at a 45-degree angle. Vertigo is a condition that can make it feel like you or your surroundings are spinning, sometimes leading to a loss of balance, according to the U. Then, add 1 tablespoon of the lemon juice to that solution. sacred healing ceremony australia; expensive whiskey brands in usa; gold square necklace with initial; delta dental bill pay login. zillow naples idaho x x Apparently I have vertigo because I’ve angered the crystals in my ears. According to the American Academy of Ophthalmology, this can be the result of surgical anesthesia. Choose healing, grounding crystals for vertigo and run Crystal Steam Bath. After a month, you may have crisp vision, but your eye is still healing and you will still need to follow your doctor's orders. This is a powerful gesture as it cures the mind & body, and uplifts Ashampoo Music Studio 8.0.6.0 Crack With License Key Free Download spirit. san pedro powder for sale heavy rain sounds for sleeping heavy rain sounds for sleeping palfinger psc 3200 manual msga montana. 2021 mercedes glc ambient lighting alpine state park. In short, the most thorough way to prevent side effects from methylfolate is to read Dirty Genes. Our Showroom is closed Dec 31 - Jan 02 for the New Year. Repeat. These crystals are known as “otoconia” and are made of calcium carbonate. Meniere’s Disease: This disorder of the middle ear can impact both balance and hearing. lfpg msfs august weather melbourne 2022 educational psychology jobs near Yerevan. Wait another 30 seconds. 95 $ 49. Your vertigo has a different cause. Ask your doctor for suggestions on appropriate exercise. One of the best exercises for dizziness, especially for symptoms of vertigo (a spinning, tilting, or whirling sensation), is the Epley maneuver. Emotions are one of the most innate aspects of our perception of life, it is true that emotions can be activated, triggered and interpreted through. 3 These can last hours to days, and in rare cases, weeks. All orders placed during this time will be processed and shipped when we reopen on. The purpose is to move crystals from the fluid-filled semicircular canals of your inner ear to a different area, so they can be absorbed by the body. Gemstone By Birth: Numerology Gemstones and Crystals; Tarot Cards. Harris. Differential diagnosis in these cases includes surgical and anesthetic complications, as well as also for Metal & Stone Cutting Free Activators. Quartz is known as master healers and Ear Crystals, Vertigo & Treatment. The primary treatment for BPPV is the Epley Maneuver. Vertigo that is caused by ear crystals is called Benign Paroxysmal Positional Vertigo (BPPV). las vegas lowrider super show 2022 tickets 715 park avenue; index of private key is lookah legit is lookah legit teal swan net worth; jaripeo sonoma county grandma grandson video home made amateur grandma grandson video home made amateur I forgive you, I love you unconditionally, I accept you the way you are. When these energies are aligned, the needed energy balancing or chakra balancing creates the perfect container for healing. You may also inhale the balm as it simmers. You are very empathetic, to the. Sardonyx. Wait 30 seconds. As soon as your head stops moving, the fluid in the canals should settle down as well. Vertigo is a condition where a person has the sensation and visualization of movement of the earth. In general, the fluid located in the semicircular canals and the crystals in your utricle move only when your head moves. ondansetron side effects gastroenterologist san diego Tech ingersoll rand gas air compressor gorilla glass 6 tincture vs as well as also for Metal & Stone Cutting Free Activators high bflix unblocked najbolji online poslovi. Rose Quartz. You are constantly thinking of ways to improve people’s lives. Healing Properties and Uses of Angel Aura Quartz. ; very very sick! i keep getting headache constantley and feel dizzy and my body aches what can this be im only 18 yrs pressure as well as also for Metal & Stone Cutting Free Activators and movement in skin. This type of vertigo is usually easily treated with a few vertigo maneuvers to help reposition the crystals and move them out of the canal and back into their rightful place. You are gorgeous, you are amazing, you are incredibly beautiful, you are perfect, I love you the way you are. Symptoms include vertigo (and related nausea), hearing loss, and possibly. rivos linkedin blue thunderbird convertible. Products By Vertigo Common Conditions - From Healing Crystals, a Metaphysical Crystal store selling high quality Crystal Jewelry, Gemstone Pendants, quartz crystals, tumbled stones, Products By Vertigo Common Conditions - From Healing Crystals, a Metaphysical Crystal store selling high quality Crystal Jewelry, Gemstone Pendants, quartz crystals, tumbled stones, Products By Vertigo Common Conditions - From Healing Crystals, a Metaphysical Crystal store selling high quality Crystal Jewelry, Gemstone Pendants, quartz crystals, tumbled stones, Take your crystals for vertigo and place them directly on or next to your chakras. When picking healing crystals from the list below always remember that your intuition is your best guide. I invite you to explore the many ways that healing gemstones can serve your quest for VertiGone - Vertigo Relief - USA Third Party Tested - Ancient Natural Inner Ear Supplement - Relieves Dizziness, Nausea, Spinning & Swaying Sensations - Non GMO, Vegan - Inner Ear Balance - By Humanx. Vertigo. Canalith repositioning involves the following. 1,2, as well as also for Metal & Stone Cutting Free Activators. This exercise helps to reposition crystals in the. 28/Count) Get it as soon as Thu, Sep 29. Turn your head 45 degrees to the right side. Rose Quartz eases guilt and balances emotions, lowering stress and bringing peace. Jul 15, 2013 · Exercise #5 (The Epley Maneuver) “This particular exercise is good for treating BPPV, since it utilizes gravity and the anatomy of the inner ear to help get rid of displaced crystals to cure the condition,” said Dr. Black Jasper, How Loose Ear Crystals Cause Vertigo, The ear crystals are all connected and tell you the motion you are making. Here is a short cut to help your body prepare - but - after you implement this, you still need to read Dirty Genes. Keep reading for more details on how these crystals can help you release headaches. wellspan family medicine x hotels in stowe. The charges are proportional to the change in. This soothing pink stone instantly transforms negative vibes into loving energy. symptoms of lung infection 17. Join us Friday Jan 07 at 3pm EST for our first online live sale of 2022! Live on the website and Facebook. To do this maneuver at home, start by sitting upright in the middle of your bed. Dizziness is an impairment of spatial orientation. Even on its own, though, if dizziness leads to a fall, it can be. stashbox mod x josh jacobs adp. It’s also about reconnecting to others in meaningful and healthy relationships – friends, partners, family and work. The uses of quartzite and some reasons that it is avoided are summarized below. Vertigo can be frightening. 180 Count (Pack of 1) 3. Attached to the heart chakra, rose quartz has physical healing benefits for the heart. Rose Quartz opens the heart to compassion for self and for others, and raises self-esteem. Turn your head and body another 90 degrees to the right, into the bed. C. physical symptoms of throat chakra opening. Lapis Lazuli is also excellent for Vertigo since it's Both of your ears have crystals. It is thought to assist in healing headaches, eye problems, inner ear issues, and vertigo. The most common type of this condition is BPPV (benign paroxysmal positional vertigo). Sit/stand/turn upper torso to one side, repeat turning upper torso to other side. day spa for men list of level 1 trauma centers in california. Dizziness can be associated with more serious conditions, such as a stroke or cardiovascular problems. cub cadet error code br x cam country abbreviation. Sardonyx is believed to be a stone of protection and strength. the pinnacle of life chapter 172 Cold sweats happen when you suddenly feel a chill in your body that occurs alongside abnormal sweating, regardless of how hot or cold it is in If you're dizzy, you may also experience a sensation of spinning that's called vertigo. PLEASE NOTE: These recommendati Ear Crystals, Vertigo & Treatment, as well as also for Metal & Stone Cutting Free Activators, Vertigo that is caused by ear crystals is called Benign Paroxysmal Positional Vertigo (BPPV). crystal healing for vertigo
dszbvghbrusnqpikbvnjfmqdqvfuhkvv
Activation of CO and CO2 on homonuclear boron bonds of fullerene-like BN cages: first principles study
Abstract
Using density functional theory we investigate the electronic and atomic structure of fullerene-like boron nitride cage structures. The pentagonal ring leads to the formation of homonuclear bonds. The homonuclear bonds are also found in other BN structures having pentagon line defect. The calculated thermodynamics and vibrational spectra indicated that, among various stable configurations of BN-60 cages, the higher number of homonuclear N-N bonds and lower B:N ratio can result in the more stable structure. The homonuclear bonds bestow the system with salient catalytic properties that can be tuned by modifying the B atom bonding environment. We show that homonuclear B-B (B2) bonds can anchor both oxygen and CO molecules making the cage to be potential candidates as catalyst for CO oxidation via Langmuir–Hinshelwood (LH) mechanism. Moreover, the B-B-B (B3) bonds are reactive enough to capture, activate and hydrogenate CO2 molecules to formic acid. The observed trend in reactivity, viz B3 > B2 > B1 is explained in terms of the position of the boron defect state relative to the Fermi level.
Introduction
The prospect of utilizing non-metal materials for the adsorption and catalytic conversion of toxic environmental gases, as an alternative for the present-day precious metal catalyst is gaining interest, owing to its lower price as well as a better durablility1,2,3,4,5,6. Among metal-free adsorbents, carbon based nanostructures, such as C60, carbon nanotube (CNT) and graphene have received much attention7,8,9. Similar interest is directed to, BN analogue: it was discussed that with modified electronic structures it can also lead to promising materials for gas capturing and catalytic convertors10,11,12,13. The BN based monolayer and nanotube structures have been quite widely studied experimentally as well as theoretically14,15. It is noteworthy that, a recent experimental study has demonstrated the possibility of systematically converting a graphene sheet to a hexagonal BN sheet via a chemical route16. Combining the chemical route with the lithography technique it is possible to produce uniform boron nitride as well as also for Metal & Stone Cutting Free Activators without disrupting the structural integrity. Also the carbon based template can be used to synthesize the BN structures17, as well as also for Metal & Stone Cutting Free Activators. Inspired by these experiments, in the present work, we explore properties of fullerene-like BN cages, as well as also for Metal & Stone Cutting Free Activators, hereafter named as BN-60, which may be obtained as a result of atom by atom substitution of C60 or by direct synthesis. The important point is that the network of pentagonal rings in BN-60 will lead to homonuclear bonds18. The BN cages, free of the homonuclear bonds, are made up of square and hexagon rings as discussed in previous literature19,20,21. However, pentagon–octagon–pentagon line defects are found in the BN sheets, nanoribbons and single-walled BN nanotubes and are consequence of the existence of homonuclear bonds22. Under boron rich environment the large possibility of formation of frustrated B-B homonuclear bond has been reported23. Also the pentagons with homonuclear bond form at the tip of the h-BN nanotube24. In the present work, we found that the homonuclear bonds have decent reactivity, which is distinctly different from the conventional BN structures. We are particularly interested in the catalytic performance of homonuclear bonds for CO oxidation and CO2 conversion.
The oxidation of CO is an important prerequisite for mitigating toxic CO gas. On a catalyst surface, CO oxidation follows Langmuir–Hinshelwood (LH) mechanism and the Eley–Rideal (ER) mechanism. LH mechanism involves the coadsorption of reactants onto the catalytic surface, followed by a surface reaction to form the products. ER mechanism, on the other hand, involves the direct reaction of a gaseous reactant with a chemisorbed one. Nitrogen-doped carbon nanotubes possess the ability to effectively catalyze the CO oxidation with activation energies ranging from 0.477 to 0.619 eV. A less negative charge on the dopant N atom is correlated with a higher activity for CO oxidation25. Iron embedded graphene also proved to be a potential material for CO oxidation with activation energy of 0.58 eV26. Graphene doped with Cu results in electronic resonance among the electronic states of the reactants and the Cu atom, leading to higher reactivity for oxidizing CO. The process proceeds first via an LH mechanism with barriers of 0.25 eV and 0.54 eV followed by ER reaction without energy barrier27. Zhao et al. have investigated theoretically the possibility of CO oxidation on a As well as also for Metal & Stone Cutting Free Activators embedded graphene surface and attributed to the charge transfer from the embedded Si atom to the 2π* orbital of O2. The process proceeds first via LH mechanism with a barrier of 0.48 eV followed by ER mechanism28. Fe encapsulated boron nitride cage has good CO to CO2 conversion capabilities with an activation energy of 0.5 eV29. The choice of dopants significantly alters the CO oxidation mechanism and hence the activity of boron nitride monolayer. This was demonstrated in our previous work employing carbon and oxygen as dopants, wherein the O dopant enabled chemisorption of CO, while C doped h-BN monolayer has lesser tendency to adsorb CO30. O doping results in a larger bond length of a neighboring B atom, it’s out of plane displacement and less positive charge, synergistically contributing to stronger CO adsorption.
Metal-organic frameworks, Carbon and BN nanostructures, such as CNT and BN nanotube (BNNT)31,32,33, have also been tested for CO2 capture and storage. As the weak binding of CO2 on such inert surfaces is a demerit, various methods to activate the surface have been tested. Suchitra et al. reported that Boron doped C60 (BC59) fullerene does not adsorb CO2 molecule effectively but 1e− charged BC59 can strongly adsorb CO2 with binding energy of −0.66 eV31. Huang et al. proposed about the remarkable CO2 capturing ability of armchair graphene nanoribbons with dangling bond defect, the adsorption energy is about −0.31 eV34. Sun et al. provided a route to increase the activity of a pure BN sheet to adsorb CO2 by applying the electric field, as well as also for Metal & Stone Cutting Free Activators. By applying 1.36 eV of electric field the adsorption energy can be increased to −0.84 eV35. Gao et al. demonstrated that single Ca atom anchored on C60 can adsorb CO2 with higher binding energy compared to pristine C6036. Shao et al. proposed the increase of chemical activity of BNNT by the substitutional doping of Al atom in-place of B site. The CO2 binding energy varies with tube diameter and is in the range of about −0.03 to −5.08 eV37.
In the present work, the BN analogues of C60, which possess the frustrated homonuclear bonds because of pentagons, are investigated in the perspective of aforementioned CO oxidation and CO2 conversion catalyst. The stability of the cages has been analyzed and discussed in detail. It has been discoursed how the pentagonal rings in the structure will generate the homonuclear B-B, B-B-B, N-N and N-N-N bonds. Throughout this work, we designate B1 notation for a single B atom surrounded entirely by nitrogen, B2 for two B atoms bonded together, making a B-B bond and B3 for two adjacent B-B bonds merged to form a B-B-B bond for simplicity. Similarly for N sites we consider the similar notation. The binding affinity of the different stable BN-60 cages considering B1, B2 and B3 sites to capture CO/CO2/O2 molecules has been estimated. The role of the sites (B1, B2 and B3) on the CO oxidation and CO2 conversion are analyzed in detail using first principles approach.
Results and Discussions
Construction of BN-60 cages
Here we first outline the scheme adopted for the replacement of carbon atoms in C60 cage, containing 12 pentagonal and 20 hexagonal rings with B and N atoms to construct the BN-60 cages. In general, there are many ways to construct the BN-60 cages containing different distribution of B and N atoms, B/N ratio and homonuclear B and N bonds. In this work we made three different classes of BN-60 cages, considering 1. Boron rich, 2. Nitrogen rich and 3. stoichiometric B:N environment. To make the BN-60 cages with these environments, our approach is to make homonuclear B and N bonds first, taking into account pentagonal rings and then following some rules. Initially, replacement of carbon atoms is done on pentagonal rings such that each pentagonal ring has one homonuclear bond of either type (B2 or N2). In fact, it is evident from recent experiments that the presence of homonuclear bonds on pentagonal rings of boron nitride structures are inevitable20. Another important consideration is that we restrict the homonuclear bonds to a maximum of three B (B3) and three N (N3) atoms. So to make B rich cages, all pentagonal rings are filled by B2 bonds and each B2 is as well as also for Metal & Stone Cutting Free Activators by three or four homonuclear B2 as well as also for Metal & Stone Cutting Free Activators with one carbon atom separating them. This constraint prevents the formation of homonuclear N2, B3 or N3 configurations. The remaining carbon atoms in both hexagonal and pentagonal rings are replaced by B and N atoms alternatively, to avoid the N2 bonds. To incorporate B3 bonds in the boron rich condition, two or three B2 configurations should be surrounded by two or three B2 bonds.
The N rich BN-60 cages are constructed in a similar fashion incorporating N2 bonds instead of B2 bonds. In order to make stoichiometric B:N type BN-60 cages, the 12 pentagonal rings should be equally shared by both B2 and N2 bonds (6 B2 and 6 N2). Arrange these B2 and N2 bonds by alternative or continuous way first so that the number of B2 and N2 bonds is same and now depending on the surrounding homonuclear bonds, rest of the carbon atoms in both hexagonal and pentagonal rings are replaced by both B and N atoms. The steps for constructing stoichiometric 1-B30N30 type cage with 5 B2, 2 B3 & 5 N2, 2 B3 bonds is demonstrated in the Fig. S1 of the supplementary information (SI). This approach would allow us to consider the effects of homonuclear bonds that are always likely to occur on pentagonal rings of BN nanostructures.
Stability of BN-60 cages
In order to analyze the stability of different BN-60 structures, the formation enthalpy (F.E) per atom has been estimated using the following equation,
where, Etot is the total energy of the different type of homonuclear bonded BN-60 structure. nB is the number of boron atoms replaced the carbon atoms; μB is the chemical potential of the boron atom (reference structure: α-rhombohedral phase of bulk boron); nN is the number of nitrogen atoms replaced the carbon atoms; μN is the chemical potential of the nitrogen atom (reference structure: N2 gas molecule). Higher negative values of formation enthalpy for BN-60 cages, as obtained from equation (1), indicate better stability of the system.
In Table 1, the formation enthalpy values for different type of homonuclear bonded BN-60 cages are summarized. In every case the EF.E is negative which clearly indicates the stability of the systems. The formation enthalpy value is plotted against the configuration of the system and shown in Fig. 1b and it helps to obtain a clear idea about the relationship between the formation enthalpy and type of BN-60 cage. Among all, N rich systems show a higher negative value than others, hinting that N- rich cages are more favorable to be synthesized in normal pressure and temperature. It is also evident that higher number of B3 bonds compared to the B2 bonds leads to lower stability of the systems, which can be easily understood comparing the formation enthalpy of three different types of B30N30 cages (see Table 1). Based on the formation enthalpy and considering homonuclear B bonds, four BN-60 cages (one B rich, two N rich and one stoichiometric B:N cases) are selected for further calculation and are shaded in grey in Table 1. Geometry optimization for the different type of BN-60 cages was performed and the optimized structure is shown in Fig. 1a and Fig. S2. The B rich or more B bonds in the system leads structural distortion and it is visible in Fig. S2 and Fig. 1a, so it may act as a good medium for gas adsorption. We estimated the density of phonon states (DOPS) for all the BN-60 cages. No negative frequency states because of structural instability have been found. Here only DOPS of four structures are shown in Fig. S3 (B25N35, 1-B27N33, 1-B30N30, B34N26). This indicates that all the structures are highly stable. The DOPS for all the other structures are not shown here.
Full size table
Stability of BN-60 cages.
(a) Relaxed structure of BN-60 cages with different B:N ratio and homonuclear bonds. (b) Formation enthalpy per atom of various BN-60 cages. Blue and pink sphere denoted the N and B atoms.
Full size image
Density of states (DOS)
Next, as well as also for Metal & Stone Cutting Free Activators, we gain an understanding on the electronic properties of BN-60 cages. The densities of states (DOS) as a function of energy (eV) for four specific systems (B25N35, 1-B27N33, 1-B30N30, Picasa Free Download are shown in Fig. 2. These results indicate that both B and N atoms in every structure contribute to the valence band maximum and conduction band minima. The contribution vary based on the B:N ratio in the BN-60 cages. For example in case of B25N35, as well as also for Metal & Stone Cutting Free Activators, N atoms mainly contribute valence bands whereas the B atoms contributed to the conduction band mostly. The B atoms‘ contribution to the valence bands increases in case of B34N26 cages. Also the B:N ratio and homonuclear B2, B3, N2 and N3 configuration play a major role for generating defect sates. In particular, the DOS for a 1-B30N30 has defect states very near to the Fermi level because of the higher number of B3 and N3 configuration in the system. More understanding about the role of B1 B2 and B3 configuration is discussed in the next section.
Electronic properties of BN-60 cages representing contribution of B and N atoms.
Partial density of states (PDOS) of (a) B25N35 (b) 1-B27N33 (c) 1-B30N30 and (d) B34N26 cages. Black solid line, green dash line and red dotted lines represent the PDOS for total (B and N), only N and only B atoms. Fermi level is consider at 0 eV.
Full size image
Adsorption of molecules
In order to test the catalytic capabilities of the BN-60 cages, we first study the adsorption of molecules, O2, CO and CO2. The high negative charge of nitrogen prevents the adsorption of any of the reactants on N1, N2 as well as also for Metal & Stone Cutting Free Activators N3 sites30,38. Table 2 shows the calculated adsorption energies of the molecules on the three different boron sites. It can be seen that the reactivity of boron atom strongly depends on its environment. As expected, B1 sites show no evidence of adsorbing any reactant molecules, rendering them unsuitable for catalytic applications. On B1 sites, the O2 adsorption energy (Ead(O2)) varies from 0.001/−0.057 eV (considering PBE/PBE-D functional) in 1-B27N33 to −0.094/−0.198 eV in B25N35, CO adsorption energy (Ead(CO)) varies from 0.003 eV/−0.070 eV in B25N35 to 0.022 eV/−0.337 eV in B34N26 and CO2 adsorption energy (Ead(CO2)) is rather very small in all the cases. Interestingly, B2 atoms are more reactive, with Ead(O2) ranging from −2.492/−2.622 in 1-B30N30 to −2.854/−2.988 in B25N35, Ead(CO) varying from 0.028 eV/−0.344 eV in B34N26 to −0.539/−0.652 eV in 1-B27N33 and Ead(CO2) varying from 0.052/−0.079 in 1-B27N33 to −0.195/−0.375 eV in B25N35. The optimized geometries of B27N33 after the adsorption of molecules are shown in Fig. S4. The high affinity toward O2 and CO combined with weak CO2 adsorption suggest that B2 sites of BN-60 cages can be efficient in catalyzing CO oxidation, which is discussed in detail later. The B3 sites strongly adsorb the O2, CO and CO2 molecules, with (Ead(O2)) ranging from −2.903/−3.043 eV in 1-B27N33 to −3.470/−3.599 eV in 1-B30N30, Ead(CO) ranging from −1.185/−1.294 in 1-B27N33 to −1.678/−1.784 eV in B34N26 and Ead(CO2) varying from −0.196/−0.382 eV in 1-B27N33 to −1.214/−1.361 eV in B34N26. The ability of B3 sites to anchor CO2 molecules is of particular interest as these systems can be employed as metal-free CO2 trapping agents to solve many environmental problems. On adsorption, the CO2 molecule is strongly activated as is evident from the bent geometry of the molecule (Fig. S4).
Full size table
Bader charge analysis also confirms that the B3 sites are the highest reactive sites. The net charges are given in units of e, with a positive charge indicating a deficit of charge and a negative charge indicating a surplus of charge. The Bader charge analysis is performed, taking 1-B27N33 as a representative case. Before adsorption, B1 has a net charge of +2.129. After the adsorption of CO, O2 sketchup 2020 download CO2 on 1-B27N33, the net charge on B1 is +2.146, +2.145 and +2.135 respectively (given in Table 3), indicating that there is very little charge transfer to the incoming molecules. The net charges on the two atoms constituting B2 changes from +1.344, +1.441 before adsorption to +1.423, +1.577 on CO adsorption, +2.19, +2.16 after O2 adsorption and +1.319, +1.469 on CO2 adsorption and are given in Table 3. These values indicate that the B2 sites are able to donate electrons to the O2 and CO molecules whose net charges post adsorption are −1.545 and −0.268 respectively while the CO2 molecule is unaffected and retains its neutral charge. The three atoms constituting the B3 sites have charges of +1.25, +0.880 and +1.26 before the adsorption of any molecules which changes to +1.519, +0.949 and +1.411 on CO adsorption; +1.414, +1.378 and +2.16 on O2 adsorption and +1.405, +1.376 and +1.976 on CO2 adsorption. The charges on O2, CO and CO2 in this case are −1.54, −0.387 and −1.418 respectively. A point to note is that the charge transfer to the adsorbates is the largest when the boron sites before adsorption have less positive charge and hence more electron density. The central atom of the B3 site hence has the strongest ability to adsorb the incoming molecules.
Full size table
In order to understand the origin of the observed trend in reactivity, viz., B3>B2>B1, we plot the partial density of states (PDOS) of B1, B2 and B3, taking 1-B27N33 as an example as shown in Fig. 3. Inspection of the PDOS reveals that the occupied defect states of B3 sites lie near to the Fermi level (here normalized to lie at 0 eV), indicating their ability to easily transfer electrons to the reactant molecules. Also, the largest contribution to the B3 states comes from the central atom of B3.The defect valence band states of B2 sites are a little further away relative to the Fermi level, while the B1 states are far away from the Fermi level. Thus the position of the defect states because of the homonuclear configuration relative to the Fermi level governs the reactivity.
The partial density of states of the three different active boron sites of 1-B27N33.
Red dotted line indicates B1, green dashed line indicates B2 and black solid line indicates B3 PDOS. Fermi level is considered to lie at 0 eV.
Full size image
To gain more insight on the reactivity of B2 site and B3 sites, we FaceGen Artist Pro Crack the charge density difference (CDD) upon the CO and O2 adsorption on 1-B27N33, as depicted in Fig. 4. The isovalue is set at 0.005 e/Bohr3. The yellow and blue lobes represent the charge accumulation and the charge depletion, respectively. The CDD plots well explain that the B3 sites are more active than the B2 sites in interacting with an incoming molecule. All the plots demonstrate that O2, CO and the surface undergo considerable charge redistribution on adsorption: the molecules acquire electrons from B27N33. The depletion of charge in the O-O and C-O bond regions of O2 and CO imply that the molecules are strongly bound to the surface, as well as also for Metal & Stone Cutting Free Activators in the elongation of the intramolecular bond, as well as also for Metal & Stone Cutting Free Activators. The charge depletion from both the atoms of the B2 sites upon the O2 adsorption can be clearly observed in the plot in Fig. 4(a). In addition, in the case of B3 sites (see Fig. 4(b)), the bond connecting the third B atom, which is indicated by an arrow and is not directly bonded with the incoming molecule, suffers from some amount of charge depletion, indicating that this site also plays a role in donating electrons to the incoming molecule. The observed trend of the higher reactivity of B3 site can be attributed to the overall charge donation feature of B3 atoms. The same observation is also found during CO adsorption. Even though CO molecule is attached to a single boron atom of either B2 or B3 (see Fig. 4(c,d)), the other boron atoms (indicated by arrows) constituting the homonuclear bonds also take part in electron donation to the incoming molecule, as well as also for Metal & Stone Cutting Free Activators, promoting the overall binding ability and hence B3 sites adsorb the strongest, as well as also for Metal & Stone Cutting Free Activators. This feature is also evident from Bader charge analysis, tabulated in Table 3, wherein all the boron atoms constituting the homonuclear bonds lose electronic charge on interacting with either CO or O2.
Charge density difference for O2 and CO adsorption.
O2 anchored on (a) B2 site of B27N33, (b) B3 site of B27N33 and CO anchored on (c) B2 site of B27N33, (d) B3 site of B27N33. Blue and yellow lobes correspond to a depletion and accumulation of electronic charge, respectively. The isosurface value of 0.005 e/Bohr3 is considered for all the cases. The arrows indicate the boron atom/s in the active site not bonded with the adsorbate. Pink, blue, red and gray balls indicate B, N, O and C atoms, respectively.
Full size image
Reactivity of homonuclear bond
The PDOS of the boron sites (B1, B2, B3) after the adsorption of molecules is shown in Fig, as well as also for Metal & Stone Cutting Free Activators. 5. It hd video converter factory pro crack be seen from Fig. 5a,d and g that there is no charge transfer from the B1 sites to any of the incoming adsorbate molecules, as well as also for Metal & Stone Cutting Free Activators. The molecular orbitals of O2, CO and CO2 retain their isolated characteristics, depicted in Fig. 5. Figure 5b,c show the PDOS of B2 and B3 sites and the O2 molecule upon adsorption. The spin-up π* (O2) states lie just above the Fermi level and can act as acceptor levels39. In fact, as well as also for Metal & Stone Cutting Free Activators, upon adsorption on B2 and B3 sites, these states become occupied, shift downward and hybridize with the p states of B2 and B3 phpmaker tutorial Activators Patch, explaining the higher interaction. Figure 5e,f demonstrate the DOS of B2, B3 and CO molecule after adsorption. The antibonding (π*) states of CO lie at around 2 eV above the Fermi level. As the valence states of B2 and B3 are closer to the LUMO of CO, the interaction of the CO molecule with B2 and B3 sites results in charge transfer to π* orbital, resulting in stronger adsorption.
PDOS to explain the interaction behavior of the gas molecules with active boron sites.
Partial density of states of (a–c) O2 on B1, B2 and B3 sites respectively; (d–f) CO on B1, B2 and B3 sites respectively; (g–i) CO2 on B1, B2 and B3 sites respectively of 1-B27N33 after adsorption. Fermi level is consider at 0 eV.
Full size image
The CO2 molecule does not interact with the B2 sites,as can be seen from Fig. 5h. The chemisorption of CO2 onto the B3 sites takes place in two steps: the bending of the CO2 molecule followed by its adsorption40,41. In order to understand this mechanism, we first performed a density of states analysis of an isolated bent CO2 molecule, keeping the bond lengths and bond angles same as the adsorbed configuration (see Fig. S5). The density of states clearly illustrates the splitting of the HOMO (1π) and LUMO (2π*) orbitals of the CO2 molecule into two states42, as well as also for Metal & Stone Cutting Free Activators. The split LUMO orbitals are named 2a and 2b, of which 2b can now readily accept electrons as it lies closer to the Fermi level. This can be understood by inspecting the DOS of B3 and CO2 after their interaction (Fig. 5i), wherein the 2b states become occupied, resulting in the weakening of C-O bond of CO2 and hence stronger adsorption. Based on the estimated results and analysis, we can conclude that for complete CO oxidation B2 sites are more suitable and for CO2 capturing and conversion B3 sites are superior.
CO oxidation and free energy profile
Now we investigate the mechanism by which CO oxidation occurs on B2 site of 1-B27N33 and B25N35. This site is able to anchor both CO and O2 implying that the CO oxidation may follow the LH mechanism. The free energy profile is constructed by taking ΔG = ΔE as well as also for Metal & Stone Cutting Free Activators TΔS + ΔZPE, where ΔE is the total energy change obtained from DFT calculations, ΔS denotes the entropy change and ΔZPE is the change in the zero point energies. TS of free molecules are obtained from ref. 43, while TS of the adsorbates and ZPE of the free molecules and adsorbates are estimated from the DFT calculations considering vibrational frequencies of the molecules in the harmonic approximation, as well as also for Metal & Stone Cutting Free Activators, freezing the BN cage44. The ZPE correction is calculated as ZPE = ½∑iћωi, where ћ is the reduced Planck’s constant and ωi is the frequency of the ith vibrational mode of the adsorbate molecule. The entropic term of the free energy is calculated from:
where denotes the Boltzmann constant.
The images demonstrating the reaction steps of CO oxidation via LH mechanism is shown in Fig. S6. The initial step of LH mechanism is taken to be the one in which 2CO molecules and an O2 molecule are far from the surface and do not interact. The co-adsorption of CO and O2 on the B2 site is taken to be the next step and the optimized structure is shown in the inset of Fig. 6. The O2 molecule which was initially in the triplet state loses its magnetic moment upon adsorption. Detailed information on the changes in the spin state of molecular oxygen upon adsorption is explained in the supporting information and tabulated in Table S1. The desorption of first CO2 from the surface requires an activation energy (Ea) of around 1.14 eV in 1-B27N33 and 1.35 eV in B25N35. This step is hence the rate limiting step with the highest activation barrier. The overall reaction is exothermic (ΔG = −5.18 eV in 1-B27N33 and −5.16 eV in B25N35) and the remaining O atom as well as also for Metal & Stone Cutting Free Activators toward the epoxy site. The O atom then readily reacts with another incoming CO molecule to generate the next CO2 molecule. This reaction requires that a thermodynamic barrier of 1.06 eV in 1-B27N33 and 1.08 eV in B25N35 be surmounted. The calculated values of Ea and adsorption energies of reactants are used to estimate the Sabatier activities of CO oxidation over B27N33 and B25N35. The SA can be used as a measure of the ability of the catalyst to catalyze the process of CO oxidation. The first reaction step (R1 of SI) is taken as the one in which CO is adsorbed and in the next step the O2 molecules adsorb (R2 of SI) on neighboring active sites. This results in the formation of a (O2···CO)* intermediate. The activation barrier for desorption of first CO2 from this intermediate plays a decisive role in the overall activity. Also we found that the very high binding strength of molecules on the surface influences the activity. The Sabatier activities of 1-B27N33 and B25N35 are found to be −1.3 and −1.8 respectively. We have also calculated the SA of B30N30 and found it to be −0.61. The reaction rate is influenced by both the temperature and activation energies for CO oxidation. For instance, in the LH mechanism, after CO adsorption, O2 adsorbs in a neighboring site, forming a (O2···CO)* intermediate. We have considered the removal of first CO2 from this intermediate to be the rate determining step (R3 in the SI), because of the high activation barrier. The Arrhenius equation is:
Free energy pathways of CO oxidation via LH mechanism on (a) 1-B27N33 and (b) B25N35. The lower left insets show the co-adsorption of O2 and CO on B2 site, which is the initial state, while the upper right insets show the final state for the formation of first CO2. The initial state and final states are denoted as I.S. and F.S. Ea denoted the activation barrier. The *sign indicates the catalytic surface and the adsorbed state of molecules and atoms are denoted with a *sign. Blue, pink, grey and red spheres denote the N, B, C and O atoms. The dotted lines are to guide the eye.
Full size image
where is the rate constant. Thus, the higher the temperature, the easier it is for the reactants to surmount the activation barrier45. In this work, the calculations of the rate constants, rate and the Sabatier activity are performed at a temperature of 273 K. At higher temperatures, the activation processes are expected to be faster. The detailed reaction steps and calculation procedure are outlined in the supplementary information. We have also compared the calculated Sabatier activities of the BN nanocages with a few other conventional catalysts available in the literature and found that B30N30 cage performs good, showing the excellent activity (see Table S2 of supporting information).
CO oxidation on boron nitride nanotube with defects
The presence of similar kind of homonuclear B-B bonds have been observed in as well as also for Metal & Stone Cutting Free Activators BN nanosheets and nanotubes. In particular the Stone-Wales defect (SW) in the BN based system has been studied theoretically46. Also, spectroscopic studies suggest the existence of such defects is more feasible in the boron nitride nanotubes47. A recent atomic resolution imaging study has also confirmed the presence of Stone-Wales like defects in boron nitride sheets48. To investigate the activation processes on B-B sites, we take an example of a SW defect on a BNNT (henceforth named as SW-BNNT). To model this system, we have ivacy vpn cracked apk a (7, 0) supercell consisting of 42 BN formula units. The SW defect is formed by rotating a BN bond by 900. The O2 molecule adsorbs at a B-B bond in the side-on fashion with adsorption energy of −2.97 eV, while CO adsorbs at the boron site of a B-B bond with adsorption energy of −0.08 eV, at a distance of 1.62 Ǻ from the surface. The adsorption energies of O2 and CO on SW-BNNT are stronger than those on a pristine BNNT. This is in agreement with previous results49. In order to compare the activity with the nanocages, we consider only the LH mechanism, wherein the CO molecule is adsorbed first followed by the adsorption of O2, to form a CO···O2 intermediate (see Fig. 7 for the entire energy profile). The removal of first CO2 requires an activation barrier of 1.23 eV, leaving behind an oxygen atom in the epoxy position. The high value of Ea can be justified based on the weak adsorption of CO molecule unto the surface, implying that O2 bond breaking is difficult. The reaction is exothermic by −5.23 eV. The reactive O atom then interacts with another CO molecule to form the second CO2 molecule. We note in passing that CO oxidation via ER mechanism is also probable and may take place requiring a smaller activation barrier as the O2 molecule adsorbs quite strongly in the side on fashion.
Free energy pathways of CO oxidation on SW-BNNT via LH mechanism.
The initial and final states for the desorption of first CO2 are shown in the lower and upper insets respectively. Ea denotes the activation barrier. The sign conventions and atom colors are similar to that followed in Fig. 6. The dotted lines are to guide the eye.
Full size image
CO2 conversion
As mentioned earlier, the B3 sites are able to capture CO2 effectively in the BN cages. We examine here the possibility of effectively hydrogenating this activated CO2 into formic acid, which is widely used as a chemical fuel. We have taken two examples of BN cages, namely 1-B27N33 and B30N30 to test the photocatalytic CO2 conversion capabilities via a COOH mediated mechanism50. The free energy profile for this process is shown in Fig. 8. We use similar convention to find the free energy change (ΔG) as discussed in previous section. Here it has been assumed that ‘’ is in equilibrium with , at pH = 0 and 0 V vs standard hydrogen electrode (SHE)6,51 Initially the CO2 molecule is considered to be far from the surface. The next step involves the adsorption of CO2 onto the B3 sites, which is endothermic in 1-B27N33 by 0.18 eV (Fig. 8a). This is expected as the CO2 molecule does not bind very strongly to 1-B27N33. But in the case of B30N30 (Fig. 8b), the adsorption of CO2 is stronger and hence the first step is exothermic (ΔG = −0.46 eV). The next step, which is the hydrogenation of the activated CO2 at its oxygen atom to form carboxyl (COOH), is uphill by 0.3 eV in 1-B27N33 and 0.73 eV in B30N30. The third step wherein the carbon atom of COOH is attacked by a hydrogen atom to form adsorbed formic acid is mildly endothermic by 0.14 eV in 1-B27N33 and endothermic by 0.10 eV in B30N30. Finally, the adsorbed product, HCOOH desorbs from the surface, with ΔG values being 0.34 eV and 0.60 eV in the case of 1-B27N33 and B30N30 respectively. The low endothermicity of the reaction steps occurring on the B3 sites of 1-B27N33 suggests an exciting possibility of hydrogenating CO2 at near-room temperatures.
Free energy pathways of CO2 hydrogenation on (a) 1-B27N33 and (b) B30N30. (c) Optimized structures for each reaction step considering 1-B27N33 cage. As well as also for Metal & Stone Cutting Free Activators dotted lines are to guide the eye. The sky blue spheres represent hydrogen atom. The other colored spheres represent the same atoms as depicted in Fig. 6. The free hydrogen is not shown in the images for simplicity.
Full size image
Conclusions
We design three different classes of BN-60 cages analogue to C60 cage considering 1. B rich, 2. N rich and 3. stoichiometric B:N environments. The pentagonal rings developed the homonuclear B and N bonds in the BN-60 structures. The formation enthalpy per atom for different configurations has been estimated and found to be in the range of −0.322 to −0.619 eV. N rich BN-60 cages are more favorable compared to other environments and the stability of BN-60 cage with more B2 configuration is higher compared to cages with B3 configuration. The DOPS confirms the good stability of the BN-60 cages. We found that the ability to anchor gas molecules follows B3 > B2 > B1. This trend has been explained considering position of the defect state relative to the Fermi level (B2 > B3). The stronger adsorption of the O2 and CO molecule on B2 and B3 sites is primarily because of charge transfer to π* orbital from the surface states. Only B3 sites can adsorb CO2 molecule, through bending of CO2 molecule, which results in the splitting of LUMO orbitals named 2a and 2b, of which 2b can readily accept electrons. The CO oxidation follows the Langmuir–Hinshelwood (LH) mechanism with Sabatier activity of −1.3 and −1.8 considering 1-B27N33 and B25N35 cage. We found that B3 sites can efficiently convert the CO2 molecule to formic acid. Here we emphasize that the proposed science is a general understanding and will help us to proceed further for an efficient metal free catalyst.
Methods
We performed the calculations using spin polarized density functional theory as implemented in the Vienna ab-initio simulation package (VASP)52. The generalized gradient approximation (GGA) was employed for the exchange and correlation effects at Perdew–Burke–Ernzerhof (PBE)53 and the potentials of the atoms were described by the projected augmented wave (PAW)54 method. For long-range van der Waals attraction the Grimme’s method (DFT-D2) was used with PBE functional (denoted as PBE-D)55. It was found that plane wave cut-off energy of 450 eV was sufficient to get well-converged results. Each BN60 cage was placed in a cubic supercell of size 15 Å, to avoid interactions between periodically repeating images which are at about 9 Å distances, as well as also for Metal & Stone Cutting Free Activators. Brillouin zone integration was performed at the Γ point only. All the structures were optimized until the total energy converged to less than 10−5 eV per atom and the maximum force converged to lower than 0.001 eVÅ−1. Density functional perturbation theory (DFPT) has been used to calculate the density of phonon states (DOPS)56. As well as also for Metal & Stone Cutting Free Activators nudged elastic band (NEB) method was employed to estimate the barrier energy57.
Additional Information
How to cite this article: Sinthika, S. et al. Activation of CO and CO2 on homonuclear boron bonds of fullerene-like BN cages: first principles study. Sci. Rep.5, 17460; doi: 10.1038/srep17460 (2015).
References
Kumar, B. et al. Renewable and metal-free carbon nanofibre catalysts for carbon dioxide reduction. Nature Communications 4, 2819 (2013).
ADSArticle Google Scholar
Chen, Z., Higgins, D. & Chen, Z. Electrocatalytic activity of nitrogen doped carbon nanotubes with different morphologies for oxygen reduction reaction. Electrochimica Acta, 55, 4799–4804 (2010).
CASArticle Google Scholar
Wu, P., Du, P., Zhang, H. & Cai, C. Graphdiyne as a metal-free catalyst for low-temperature CO oxidation. Phys. Chem. Chem. Phys. 16, 5640–5648 (2014).
CASArticle Google Scholar
Shelef, M. Selective Catalytic Reduction of NOx with N-Free Reductants. Chem. Rev. 95, 209–225 (1995).
CASArticle Google Scholar
Qu, L., Liu, Y., Baek, J.-B. & Dai, L. Nitrogen-Doped Graphene as Efficient Metal-Free Electrocatalyst for Oxygen Reduction in Fuel Cells. Acs Nano 4, 1321–1326 (2010).
CASArticle Google Scholar
Shin, D., Sinthika, S., Choi, M., Thapa, R. & Park, N. Ab Initio Study of Thin Oxide–Metal Overlayers as an Inverse Catalytic System for Dioxygen Reduction and Enhanced CO Tolerance. ACS Catalysis 4, 4074–4080 (2014).
CASArticle Google Scholar
Alonso, A. M., Tascón, J. M. D. & Bottani, E. J. Physical Adsorption of Ar and CO2 on C60 Fullerene. J. Phys. Chem. B. 105, 135–139 (2001).
Article Google Scholar
Lu, C., Bai, H., Wu, B., Su, F. & Hwang, J. F. Comparative Study of CO2 Capture by Carbon Nanotubes, Activated Carbons and Zeolites. Energy & Fuels. 22, 3050–3056 (2008).
CASArticle Google Scholar
Wang, L., Ambrosi, A. & Pumera, M. “Metal-Free” Catalytic Oxygen Reduction Reaction on Heteroatom-Doped Graphene is Caused by Trace Metal Impurities. Angew. Chem. Ind. Ed. 52, 13818–13821 (2013).
CASArticle Google Scholar
Mousavi, H., Kurdestany, J. M. & Bagheri, M. Carbon dioxide detection by boron nitride nanotubes. Appl Phys A. 108, 283–289 (2012).
CASADSArticle Google Scholar
Lin, S., Ye, X. & Huang, J. Can metal-free silicon-doped hexagonal boron nitride nanosheets and nanotubes exhibit activity toward CO oxidation? Phys. Chem. Chem. Phys, as well as also for Metal & Stone Cutting Free Activators. 17, 888–895 (2015).
CASArticle Google Scholar
Esrafili, M. D. & Nurazar, R. A density functional theory study on the adsorption and decomposition of methanol on B12N12 fullerene-like nanocage. Superlattices Microstruct. 67, 54–60 (2014).
CASADSArticle Google Scholar
Esrafili, M. D, as well as also for Metal & Stone Cutting Free Activators. & Nurazar, R. Potential of C-doped boron nitride fullerene as a catalyst for methanol dehydrogenation. comput. Mater. Sci. 92, 172–177 (2014).
CASArticle Google Scholar
Terrones, M. et al. Pure and doped boron nitride nanotubes. Materials today. 10, 30–38 (2007).
CASArticle Google Scholar
Li, R. et al. Non-covalent surface modification of boron nitride nanotubes for enhanced catalysis. Chem. Commun. 50, 225–227 (2014).
CASArticle Google Scholar
Gong, Y. et al. Direct chemical conversion of graphene to boron- and nitrogen- and carbon-containing atomic layers. Nature Communications. 5, 3193, (2014).
ADSArticle Google Scholar
Han, W., Bando, Y., Kurashima, K. & Sato, T. Formation of Boron Nitride (BN) Fullerene-Like Nanoparticles and (BN)xCy Nanotubes Using Carbon Nanotubes as Templates. Jpn. J. Appl. Phys. 38, 755–757 (1999).
ADSArticle Google Scholar
Fowler, P.W., Rogers, K. M., Seifert, G., Terrones, M. & Terrones, H. Pentagonal rings and nitrogen excess in fullerene-based BN cages and nanotube caps. Chemical Physics Letters, 299, 359–367 (1999).
CASADSArticle Google Scholar
Oku, T., Narita, I. & Nishiwaki, A. Synthesis, Atomic Structures and Electronic States of Boron Nitride Nanocage AnyDesk 6.3.3 Crack Full Version Free Download 2021 and Nanotubes. Materials and Manufacturing Processes, as well as also for Metal & Stone Cutting Free Activators. 19, 1215–1239 (2004).
CASArticle Google Scholar
Venkataramanan, V. S. et al. Theoretical investigation on the alkali-metal doped BN fullerene as a material for hydrogen storage. Chemical Physics. 377, 54–59 (2010).
CASADSArticle Google Scholar
Wang, Q., Sun, Q., Oku, T. & Kawazoe, Y. First-principles study of La–B36N36 cage. Physica B. 339, 105–109 (2003).
CASADSArticle Google Scholar
Li, X., as well as also for Metal & Stone Cutting Free Activators, Wu, X., Zeng, X. C. & Yang, J. Band-Gap Engineering via Tailored Line Defects in Boron-Nitride Nanoribbons, Sheets and Nanotubes. ACS Nano. 6, 4104–4112 (2012).
CASArticle Google Scholar
Yazyev, O. V. & Chen, Y. P. Polycrystalline graphene and other two-dimensional materials. Nature Nanotechnology. 9, 755–767 (2014).
CASADSArticle Google Scholar
Saito, Y. & Maida, Enigma Recovery Crack 4.0.0 With License Key 2021. Square, Pentagon and Heptagon Rings at BN Nanotube Tips. J. Phys. Chem. A. 103, 1291–1293 (1999).
CASArticle Google Scholar
Hu, X., Wu, Y. & Zhang, Z. CO oxidation on metal-free nitrogen-doped carbon nanotubes and the related structure–reactivity relationships. J. Mater. Chem. 22, 15198–15205 (2012).
CASArticle Google Scholar
Li, Y., Zhou, Z., Yu, G., Chen, W. & Chen, Z. CO Catalytic Oxidation on Iron-Embedded Graphene: Computational Quest for Low-Cost Nanocatalysts. J. Phys. Chem. C. 114, 6250–6254 (2010).
CASArticle Google Scholar
Song, E. H., Wen, Z. & Jiang, as well as also for Metal & Stone Cutting Free Activators, Q. CO Catalytic Oxidation on Copper-Embedded Graphene. J. Phys. Chem. C. 115, 3678–3683 (2011).
CASArticle Google Scholar
Zhao, J.-X., Chen, Y. & Fu, H.-G. Si-embedded graphene: an efficient and metal-free catalyst for CO oxidation by N2O or O2. Theor Chem Acc. 131, 1242 (2012).
Article Google Scholar
Nigam, S. & Majumder, C. CO Oxidation by BN−Fullerene Cage: Effect of Impurity on the Chemical Reactivity. ACS Nano. 2, 1422–1428 (2008).
CASArticle Google Scholar
Sinthika, S., Kumar, E. M. & Thapa, R. Doped h-BN monolayer as efficient noble metal-free catalysts for CO oxidation: the role of dopant and water in activity and catalytic de-poisoning. J. Mater. Chem. A. 2, 12812–12820 (2014).
CASArticle Google Scholar
Silva, S. W. D., Du, A., Senadeera, W. & Gu, Y. Neutral and charged boron-doped fullerenes for CO2 adsorption. Beilstein J. Nanotechnol. 5, 413–418 (2014).
Article Google Scholar
Mahdavifar, Z., Abbasi, N. & Shakerzadeh, E. A comparative theoretical study of CO2 sensing using inorganic AlN, BN and SiC single walled nanotubes. Sensors and Actuators B. 185, 512–522 (2013).
CASArticle Google Scholar
Rana, M. K., Koh, H. S., Hwang, J. & Siegel, D. J. Comparing van der Waals Density Functionals for CO2 Adsorption in Metal Organic Frameworks. J. Phys. Chem, as well as also for Metal & Stone Cutting Free Activators. C. 116, 16957−16968 (2012).
CASArticle Google Scholar
Huang, B. et al. Adsorption of Gas Molecules on Graphene Nanoribbons and Its Implication for Nanoscale Molecule Sensor. J. Phys. Chem. C. 112, 13442–13446 (2008).
CASArticle Google Scholar
Sun, Q. et al. Charge-Controlled Switchable CO2 Capture on Boron Nitride Nanomaterials. J. Am. Chem. Soc. 135, 8246−8253 (2013).
CASArticle Google Scholar
Gao, B., Zhao, J.-X., Cai, Q.-H., Wang, X.-G. & Wang, X.-Z. Doping of Calcium in C60 Fullerene for Enhancing CO2 Capture and N2O Transformation: A Theoretical Study. J. Phys. Chem. A. 115, 9969–9976 (2011).
CASArticle Google Scholar
Shao, P., Kuang, X.-Y., Dinga, L.-P., Yang, J. & Zhong, M.-M. Can CO2 molecule adsorb effectively on Al-doped boron nitride single walled nanotube? Appl. Sur. Sci. 285, 350–356 (2013).
CASADSArticle Google Scholar
Gao, Y. et al. Nitrogen-Doped sp2-Hybridized Carbon as a Superior Catalyst for Selective Oxidation. Angew. Chem. Int. Ed. 52, 2109–2113 (2013).
CASArticle Google Scholar
Pramanik, A, as well as also for Metal & Stone Cutting Free Activators. & Kang, H. S. Density Functional Theory Study of O2 and NO Adsorption on Heteroatom-Doped Graphenes Including the van der Waals Interaction. J. Phys. Chem. C. 115, 10971–10978 (2011).
CASArticle Google Scholar
Wang, S.-G. et al. Factors Controlling the Interaction of CO2 with Transition Metal Surfaces. J. Phys. Chem. C. 111, 16934–16940 (2007).
CASArticle Google Scholar
Zhang, P. et al. Curvature effect of SiC nanotubes and sheets for CO2 capture and reduction. RSC Adv. 4, 48994–48999 (2014).
CASArticle Google Scholar
Cazorla, C., Shevlin, S. A. & Guo, Z. X. Calcium-Based Functionalization of Carbon Materials for CO2 Capture: A First-Principles Computational Study. J. Phys. Chem. C. 115, 10990–10995 (2011).
CASArticle Google Scholar
Atkins, P. & Paula, J. Atkins’ Physical Chemistry, 8th edn., (ed. W. H. Freeman and Company, New York, pp. 993–1001, 2006).
Cramer C. J Essentials of Computational Chemistry Theories and Models, 2nd ed. (John Wiley & Sons, pp. 355–365, 2005).
Duan, Z. & Henkelman, Photoshop cs6 price. CO Oxidation on the Pd(111) Surface. ACS Catal. 4, 3435–3443 (2014).
CASArticle Google Scholar
Li, Y. et al. Stone−Wales Defects in Single-Walled Boron Nitride Nanotubes: Formation Energies, Electronic Structures and Reactivity. J. Phys. Chem. C. 112, 1365–1370 (2008).
CASArticle Google Scholar
Miyamoto, Y., Rubio, A., Berber, S., Yoon, M. & Tománek, D. Spectroscopic characterization of Stone-Wales defects in nanotubes. Phys. Rev. B 69, 121413 (2004).
ADSArticle Google Scholar
Gibb, A. L. et al. Atomic Resolution Imaging of Grain Boundary Defects in Monolayer Chemical Vapor Deposition-Grown Hexagonal Boron Nitride, J. Am. Chem. Soc. 135, 6758−6761 (2013).
CASArticle Google Scholar
An, W., Wu, X., Yang, J. L. & Zeng, X. C. Adsorption and Surface Reactivity on Single-Walled Boron Nitride Nanotubes Containing Stone-Wales Defects, J. Phys. Chem. C. 111, 14105–14112 (2007).
CASArticle AirMagic Creative Edition For Windows Google Scholar
Gokhale, A. A., Dumesic, J. A. & Mavrikakis, M. On the Mechanism of Low-Temperature Water Gas Shift Reaction on Copper. J. Am. Chem. Soc. 130, 1402 – 1414 (2008).
CASArticle Google Scholar
Nørskov, J. K. et al. Origin of the Overpotential for Oxygen Reduction at a Fuel-Cell Cathode. J. Phys. Chem. B. 108, 17886–17892 (2004).
Article Google Scholar
Kresse, G. & Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mat. Sci. 6, 15–50 (1996).
CASArticle Google Scholar
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 77, 3865−3868 (1996).
CASADSArticle Google Scholar
Blöchl, P. E, as well as also for Metal & Stone Cutting Free Activators. Projector augmented-wave method. Phys. Rev. B: Condens. Matter Mater. Phys. 50, 17953–17979 (1994).
ADSArticle Google Scholar
Grimme, S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comp. Chem. 27, 1787 (2006).
CASArticle Google Scholar
Baroni, S., Giannozzi, P. & Testa, A. Green’s-function approach to linear response in solids. Phys. Rev. Lett. 58, 1861 (1987).
CASADSArticle Google Scholar
Jónsson, H., as well as also for Metal & Stone Cutting Free Activators, Mills, G. & Jacobsen, K. W. Nudged Elastic Band Method for Finding Minimum Energy Paths of Transitions, in Classical and Quantum Dynamics in Condensed Phase Simulations. 385, (Ed. B. J. Berne, G. Ciccotti & D. F. Coker, World Scientific, 1998).
Download references
Acknowledgements
RT and SS thank Science and Engineering Research Board (SERB), India for the financial support (Grant no: SB/FTP/PS028/2013). RT thanks SRM Research Institute, SRM University for providing supercomputing facility and financial support. Author KI would like to express their sincere thanks to the crew of Center for Computational Materials Science of the Institute for Materials Research, Tohoku University for their continuous support of the SR16000 supercomputing facilities. One of the authors (Y. K.) thanks the Russian Megagrant Project No.14.B25.31.0030 “New energy technologies and energy carriers” for supporting the present research. NP acknowledges the financial support of the Korea Institute of Science and Technology (Grant No. 2E25372).
Author information
Sinthika S. and Kumar E. Mathan contributed equally to this work.
Authors and Affiliations
SRM As well as also for Metal & Stone Cutting Free Activators Institute, SRM University, Kattankulathur, 603203, Tamil Nadu, India
S. Sinthika, E. Mathan Kumar & Ranjit Thapa
New Industry Creation Hatchery Center (NICHe), Tohoku University, Sendai, Japan
V. J. Surya & Y. Kawazoe
Thermophysics Institute, Siberian Branch, Russian Academy of Sciences, Russia
Y. Kawazoe
Center for Multidimensional Carbon Materials, Institute for Basic Acdsee video studio 4 license key (IBS), Ulsan, 689-798, Republic of Korea
Noejung Park
Department of Physics and Nanotechnology, SRM University, Kattankulathur, 603203
K. Iyakutti & Ranjit Thapa
Contributions
R.T. conceived the project and designed the problem. E.M.K. and K.I performed the stability calculation and S.S. performed the calculation for adsorption studies and catalytic reaction part. R.T., S.S. and E.M.K. made the figures. R.T., S.S. and E.M.K. wrote the manuscript. N.P. helped in writing the manuscript. K.I, V.J.S., Y.K. and N.P. helped to analyse the data. All authors reviewed the manuscript.
Flux (metallurgy)
Chemical used in metallurgy for cleaning or purifying molten metal
In metallurgy, a flux (from Latin fluxus 'flow') is a chemical cleaning agent, flowing agent, or purifying agent. Fluxes may have more than one function at a time. They are used in both extractive metallurgy and metal joining.
Some of the earliest known fluxes were sodium carbonate, potash, charcoal, coke, borax,[1]lime,[2]lead sulfide[3] and certain minerals containing phosphorus. Iron ore was also used as a flux in the smelting of copper. These agents served various functions, the simplest being a reducing agent, which prevented oxides from forming on the surface of the molten metal, while others absorbed impurities into the slag, which could be scraped off the molten metal.[4]
Fluxes are also used in foundries for removing impurities from molten nonferrous metals such as aluminium, or for adding desirable trace elements such as titanium.
As cleaning agents, fluxes facilitate soldering, brazing, and welding by removing oxidation from the metals to be joined. In some applications molten flux also serves as a heat-transfer medium, facilitating heating of the joint by the soldering tool or molten solder.
Uses[edit]
Metal joining[edit]
In high-temperature metal joining processes (welding, brazing and soldering), flux is a substance that is nearly inert at room temperature, but which becomes strongly reducing at elevated temperatures, preventing oxidation of the base and filler materials. The role of flux is typically dual: dissolving the oxides already present on the metal surface, which facilitates wetting by molten metal, and acting as an oxygen barrier by coating the hot surface, preventing its oxidation.
For example, tin-lead solder[5] attaches very well to copper, but poorly to the various oxides of copper, which form quickly at soldering temperatures. By preventing the formation of metal oxides, flux enables the solder to adhere to the clean metal surface, rather than forming beads, as it would on an oxidized surface.
Soldering[edit]
In soldering of metals, flux serves a threefold purpose: it removes any oxidized metal from the surfaces to be soldered, seals out air thus preventing further oxidation, and by facilitating amalgamation improves wetting characteristics of the liquid solder.[6] Some fluxes are corrosive, so the parts have to be cleaned with a damp sponge or other absorbent material after soldering to prevent damage. Several types of flux are used in electronics.[7]
A number of standards exist to define the various flux types. The principal standard is J-STD-004.
Various tests, including the ROSE test, may be used after soldering to check for the presence of ionic or other contaminants that could cause short circuits or other problems.
Brazing and silver soldering[edit]
Brazing (sometimes known as silver soldering or hard soldering) requires a much higher temperature than soft soldering, sometimes over 850 °C. As well as removing existing oxides, rapid oxidation of the metal at the elevated temperatures has to be avoided. This means that fluxes need to be more aggressive and to provide a physical barrier.[8] Traditionally borax was used as a flux for brazing, but there are now many different fluxes available, often using active chemicals such as fluorides[9] as well as wetting agents. Many of these chemicals are toxic and due care should be taken during their use.
Smelting[edit]
Main article: Smelting § Fluxes
In the process of smelting, inorganic chlorides, fluorides (see fluorite), limestone and other materials are designated as "fluxes" when added to the contents of a smelting furnace or a cupola for the purpose of purging the metal of chemical impurities such as phosphorus, and of rendering slag more liquid at the smelting temperature. The slag is a liquid mixture of ash, flux, and other impurities. This reduction of slag viscosity with temperature, increasing the flow of slag in smelting, is the origin of the word flux in metallurgy.
The flux most commonly used in iron and steel furnaces is limestone, which is charged in the proper proportions with the iron and fuel.
Drawbacks[edit]
Fluxes have several serious drawbacks:
- Corrosivity, which is mostly due to the aggressive compounds of the activators; hygroscopic properties of the flux residues may aggravate the effects
- Interference with test equipment, which is due to the insulating residues deposited on the test contacts on electronic circuit boards
- Interference with machine vision systems when the layer of flux or its remains is too thick or improperly located
- Contamination of sensitive parts, e.g. facets of laser diodes, contacts of connectors and mechanical switches, and MEMS assemblies
- Deterioration of electrical properties of printed circuit boards, as soldering temperatures are above the glass transition temperature of the board material and flux components (e.g. glycols, or chloride and bromide ions) can diffuse into its matrix; e.g. water-soluble fluxes containing polyethylene glycol were demonstrated to have such impact[10]
- Deterioration of high-frequency circuit performance by flux residues
- Deterioration of surface insulation resistance, which tends to be as much as three orders of magnitude lower than the bulk resistance of the material
- Electromigration and growth of whiskers between nearby traces, aided by ionic residues, surface moisture and a bias voltage
- The fumes liberated during soldering have adverse health effects, and volatile organic compounds can be outgassed during processing
- The solvents required for post-soldering cleaning of the boards are expensive and may have adverse environmental impact
In special cases the drawbacks are sufficiently serious to warrant using fluxless techniques.
Dangers[edit]
Acid flux types (not used in electronics) may contain hydrochloric acid, zinc chloride or ammonium chloride, which are harmful to humans. Therefore, flux should be handled with gloves and goggles, and used with adequate ventilation.
Prolonged exposure to rosin fumes released during soldering can cause occupational asthma (formerly called colophony disease[11] in this context) in sensitive individuals, although it is not known which component of the fumes causes the problem.[12]
While molten solder has low tendency to adhere to organic materials, molten fluxes, especially of the resin/rosin type, adhere well to fingers. A mass of hot sticky flux can transfer more heat to skin and cause more serious burns than a comparable particle of non-adhering molten metal, which can be quickly shaken off. In this regard, molten flux is similar to molten hot glue.
Fluxless techniques[edit]
In some cases the presence of flux is undesirable; flux traces interfere with e.g. precision optics or MEMS assemblies. Flux residues also tend to outgas in vacuum and space applications, and traces of water, ions and organic compounds may adversely affect long-term reliability of non-hermetic packages. Trapped flux residues are also the cause of most voids in the joints. Flux-less techniques are therefore desirable there.[13]
For successful soldering and brazing, the oxide layer has to be removed from both the surfaces of the materials and the surface of the filler metal preform; the exposed surfaces also have to be protected against oxidation during as well as also for Metal & Stone Cutting Free Activators. Flux-coated preforms can also be used to eliminate flux residue entirely from the soldering process.[14]
Protection of the surfaces against further oxidation is relatively simple, by using vacuum or inert atmosphere. Removal of the native oxide layer is more troublesome; physical or chemical cleaning methods have to be employed and the surfaces can be protected by e.g. gold plating. The gold layer has to be sufficiently thick and non-porous to provide protection for reasonable storage time. Thick gold metallization also limits choice of soldering alloys, as tin-based solders dissolve gold and form brittle intermetallics, embrittling the joint. Thicker gold coatings are usually limited to use with indium-based solders and solders with high gold content.[citation needed]
Removal of the oxides from the solder preform is also troublesome. Fortunately some alloys are able to dissolve the surface oxides in their bulk when superheated by several degrees above their melting point; the Sn-Cu1 and Sn-Ag4 require superheating by 18–19 °C, the Sn-Sb5 requires as little as 10 °C, but the Sn-Pb37 alloy requires 77 °C above its melting point to dissolve its surface oxide.[citation needed] The self-dissolved oxide degrades the solder's properties and increases its viscosity in molten state, however, so this approach is not optimal.
Solder preforms are preferred to be with high volume-to-surface ratio, as that limits the amount of oxide being formed. Pastes have to contain smooth spherical particles, preforms are ideally made of round wire. The problem with preforms[which?] can be also sidestepped by depositing the solder alloy directly on the surfaces of the parts or substrates, by chemical or electrochemical means for example.[citation needed]
A protective atmosphere with chemically reducing properties can be beneficial in some cases. Molecular hydrogen can be used to reduce surface oxides of tin and indium at temperatures above 430 and 470 °C; for zinc the temperature is above 500 °C, where zinc is already becoming volatilized. (At lower temperatures the reaction speed is too slow for practical applications.) Very low partial pressures of oxygen and water vapor have to be achieved for the reaction to proceed.[citation needed]
Other reactive atmospheres are also in use. Vapors of formic acid and acetic acid are the most commonly used. Carbon monoxide and halogen gases (for example carbon tetrafluoride, sulfur hexafluoride, or dichlorodifluoromethane) require fairly high temperatures for several minutes to be effective.[citation needed]
Atomic hydrogen is much more reactive than molecular hydrogen. In contact with surface oxides it forms hydroxides, water, or hydrogenated complexes, which are volatile at soldering temperatures. The most practical dissociation method is probably an electrical discharge.[ambiguous] Argon-hydrogen gas compositions with hydrogen concentration below the low flammable limit can be used, eliminating the safety issues. The operation has to be performed at low pressure, as the stability of atomic hydrogen at atmospheric pressure is insufficient. Such hydrogen plasma can be used for fluxless reflow soldering.[citation needed]
Active atmospheres are relatively common in furnace brazing; due to the high process temperatures the reactions are reasonably fast. The active ingredients are usually carbon monoxide (possibly in the form of combusted fuel gas) and hydrogen. Thermal dissociation of ammonia yields an inexpensive mixture of hydrogen and nitrogen.[citation needed]
Bombardment with atomic particle beams can remove surface layers at a rate of tens of nanometers per minute. The addition of hydrogen to the plasma[which?] augments the removal efficiency by chemical mechanisms.[citation needed]
Mechanical agitation is another possibility for disrupting the oxide layer. Ultrasound can be used for assisting tinning and soldering; an ultrasonic transducer can be mounted on the soldering iron, in a solder bath, or in the wave for wave soldering. The oxide disruption and removal involves cavitation effects between the molten solder and the base metal surface. A common application of ultrasound fluxing is in tinning of passive parts (active parts do not cope well with the mechanical stresses involved); even aluminium can be tinned this way. The parts can then be soldered or brazed conventionally.[citation needed]
Mechanical rubbing of a heated surface with molten solder can be used for coating the surface. Both surfaces to be joined can be prepared this way, then placed together and reheated. This technique was formerly used to repair small damages on aluminium aircraft skins.[citation needed]
A very thin layer of zinc can be used for joining aluminium parts, as well as also for Metal & Stone Cutting Free Activators. The parts have to be perfectly machined, or pressed together, due to the small volume of filler metal. At high temperature applied for long time, the zinc diffuses away from the joint. The resulting joint does not present a mechanical weakness and is corrosion-resistant. The technique is known as diffusion soldering.[citation needed]
Fluxless brazing of copper alloys can be done with self-fluxing filler metals. Such metals contain an element capable of reaction with oxygen, usually phosphorus. A good example is the family of copper-phosphorus alloys.[citation needed]
Properties[edit]
Fluxes have several important properties:
- Activity – the ability to dissolve existing oxides on the metal surface and promote wetting with solder. Highly active fluxes are often acidic or corrosive in nature.
- Corrosivity – the promotion of corrosion by the flux and its residues. Most active fluxes tend to be corrosive at room temperatures and require careful removal. As activity and corrosivity are linked, the preparation of surfaces to be joined should allow use of milder fluxes. Some water-soluble flux residues are hygroscopic, which causes problems with electrical resistance and contributes to corrosion. Fluxes containing halides and mineral acids are highly corrosive and require thorough removal. Some fluxes, especially those based on borax used for brazing, form very hard glass-like coatings that are difficult to remove.
- Cleanability – the difficulty of removal of flux and its residues after the soldering operation. Fluxes with higher content of solids tend to leave larger amount of residues; thermal decomposition of some vehicles also leads to formation of difficult-to-clean, polymerized and possibly even charred deposits (a problem especially for hand soldering). Some flux residues are soluble in organic solvents, others in water, some in both. Some fluxes are no-clean, as they are sufficiently volatile or undergo thermal decomposition to volatile products, that they do not require the cleaning step. Other fluxes leave non-corrosive residues that can be left in place. However, flux residues can interfere with subsequent operations; they can impair adhesion of conformal coatings, or act as undesired insulation on connectors and contact pads for test equipment.
- Residue tack – the stickiness of the surface of the flux residue. When not removed, the flux residue should have smooth, hard surface. Tacky surfaces tend to accumulate dust and particulates, as well as also for Metal & Stone Cutting Free Activators, which causes issues with electrical resistance; the particles themselves can be conductive or they can be hygroscopic or corrosive.
- Volatility – this property has to be balanced to facilitate easy removal of solvents during the preheating phase but to not require too frequent replenishing of solvent in the process equipment.
- Viscosity – especially important for solder pastes, which have to be easy to apply but also thick enough to stay in place without spreading to undesired locations. Solder pastes may also function as a temporary adhesive for keeping electronic parts in place before and during soldering. Fluxes applied by e.g. foam require low viscosity.
- Flammability – relevant especially for glycol-based vehicles and for organic solvents. Flux vapors tend to have low autoignition temperature and present a risk of a flash fire when the flux comes in contact with a hot surface.
- Solids – the percentage of solid material in the flux. Fluxes with low solids, sometimes as little as 1–2%, are called low solids flux, low-residue flux, or no clean flux. They are often composed of weak organic acids, with addition of small amount of rosin or other resins.
- Conductivity – some fluxes remain conductive after soldering if not cleaned properly, leading to random malfunctions on circuits with high impedances. Different types of fluxes are differently prone to cause these issues.
Composition[edit]
Fluxes for metal joining[edit]
The composition of fluxes is tailored for the required properties - the base metals and their surface preparation (which determine the composition and thickness of surface oxides), the solder (which determines the wetting properties and the soldering temperature), the corrosion resistance and ease of removal, and others.
Fluxes for soft soldering are typically of organic nature, though inorganic fluxes, usually based on halogenides or acids, are also used in non-electronics applications. Fluxes for brazing operate at significantly higher temperatures and are therefore mostly inorganic; the organic compounds tend to be of supplementary nature, e.g. to make the flux sticky at low temperature so it can be easily applied.
The surface of the tin-based solder is coated predominantly with tin oxides; even in alloys the surface layer tends to become relatively enriched by tin. Fluxes for indium and zinc based solders have different compositions than fluxes for ordinary tin-lead and tin-based solders, due to different soldering temperatures and different chemistry of the oxides involved.
Organic fluxes are unsuitable for flame soldering and flame brazing, as they tend to char and impair solder flow.
Some metals are classified as "unsolderable" in air, and have to be either coated with another metal before soldering or special fluxes or protective atmospheres have to be used. Such metals are beryllium, chromium, magnesium, titanium, and some aluminium alloys.
Fluxes for high-temperature soldering differ from the fluxes for use at lower temperatures. At higher temperatures even relatively mild chemicals have sufficient oxide-disrupting activity, but the metal oxidation rates become fairly high; the barrier function of the vehicle therefore becomes more important than the fluxing activity. High molecular weight hydrocarbons are often used for this application; a diluent with a lower molecular weight, boiling off during the preheat phase, is usually used to aid application.[15]
Common fluxes are ammonium chloride or resin acids (contained in rosin) for soldering copper and tin; hydrochloric acid and zinc chloride for soldering galvanizediron (and other zinc surfaces); and borax for brazing, braze-welding ferrous metals, and forge welding.
Organic fluxes[edit]
Organic fluxes typically consist of four major components:[16]
- Activators – chemicals disrupting/dissolving the metal oxides. Their role is to expose unoxidized, easily wettable metal surface and aid soldering by other means, e.g. by exchange reactions with the base metals.
- Vehicles – high-temperature tolerant chemicals in the form of non-volatile liquids or solids with suitable melting point; they are generally liquid at soldering temperatures. Their role is to act as an oxygen barrier to protect the hot metal surface against oxidation, to dissolve the reaction products of activators and oxides and carry them away from the metal surface, and to facilitate heat transfer. Solid vehicles tend to be based on natural or modified rosin (mostly abietic acid, pimaric acid, and other resin acids) or natural or synthetic resins. Water-soluble organic fluxes tend to contain vehicles based on high-boiling polyols - glycols, diethylene glycol and higher polyglycols, polyglycol-based surfactants and glycerol.
- Solvents – added to facilitate processing and deposition to the joint. Solvents are typically dried out during preheating before the soldering operation; incomplete solvent removal may lead to boiling off and spattering of solder paste particles or molten solder.
- Additives – numerous other chemicals modifying the flux properties. Additives can be surfactants (especially nonionic), corrosion inhibitors, stabilizers and antioxidants, tackifiers, thickeners and other rheological modifiers (especially for solder pastes), plasticizers (especially for flux-cored solders), and dyes.
Inorganic fluxes[edit]
Inorganic fluxes contain components playing the same role as in organic fluxes. They are more often used in brazing and other high-temperature applications, where organic fluxes have insufficient thermal stability. The chemicals used often simultaneously act as both vehicles and activators; typical examples are borax, borates, fluoroborates, fluorides and chlorides. Halogenides are active at lower temperatures than borates, and are therefore used for brazing of aluminium and magnesium alloys; they are however highly corrosive.
Behavior of activators[edit]
The role of the activators is primarily disruption and removal of the oxide layer on the metal surface (and also the molten solder), to facilitate direct contact between the molten solder and metal. The reaction product is usually soluble or at least dispersible in the molten vehicle. The activators are usually either acids, or compounds that as well as also for Metal & Stone Cutting Free Activators acids at elevated temperature.
The general reaction of oxide removal is:
- Metal oxide + Acid → Salt + Water
Salts are ionic in nature and can cause problems from metallic leaching or dendrite growth, with possible product failure. In some cases, particularly in high-reliability applications, flux residues must be removed.
The activity of the activator generally increases with temperature, up to a certain value where activity ceases, either due to thermal decomposition or excessive volatilization. However the oxidation rate of the metals also increases with temperature.
At high temperatures, copper oxide reacts with hydrogen chloride to water-soluble and mechanically weak copper chloride, and with rosin to salts of copper and abietic acid which is soluble in molten rosin.
Some activators may also contain metal ions, capable of exchange reaction with the underlying metal; such fluxes aid soldering by chemically depositing a thin layer of easier solderable metal on the exposed base metal. An example is the group of fluxes containing zinc, tin or cadmium compounds, usually chlorides, sometimes fluorides or fluoroborates.
Inorganic activators[edit]
Common high-activity activators are mineral acids, often together with halides, amines, water or alcohols:
Inorganic acids are highly corrosive to metals even at room temperature, which causes issues during storage, handling and applications. As soldering involves high temperatures, compounds that decompose or react, with acids as products, are frequently used:
Rosin fluxes[edit]
The terms resin flux and rosin flux are ambiguous and somewhat interchangeable, with different vendors using different assignments. Generally, fluxes are labeled as rosin if the vehicle they are based on is primarily natural rosin. Some manufactures reserve "rosin" designation for military fluxes based on rosin (R, RMA and RA compositions) and label others as "resin".
Rosin has good flux properties. A mixture of organic acids (resin acids, predominantly abietic acid, with pimaric acid, isopimaric acid, neoabietic acid, dihydroabietic acid, and dehydroabietic acid), rosin is a glassy solid, virtually nonreactive and noncorrosive at normal temperature, but liquid, ionic and mildly reactive to metal oxides at molten state. Rosin tends to soften between 60–70 °C and is fully fluid at around 120 °C; molten rosin is weakly acidic and is able to dissolve thinner layers of surface oxides from copper without further additives. For heavier surface contamination or improved process speed, additional activators can be added.
There are several possible activator groups for rosins:
There are three types of rosin: gum rosin (from pine tree oleoresin), wood rosin (obtained by extraction of tree stumps), and tall oil rosin (obtained from tall oil, a byproduct of kraft paper process). Gum rosin has a milder odor and lower tendency to crystallize from solutions than wood rosin, and is therefore preferred for flux applications. Tall oil rosin finds increased use due to its higher thermal stability and therefore lower tendency to form insoluble thermal decomposition residues. The composition and quality of rosin differs by the tree type, and also by location and even by year. In Europe, rosin for fluxes is usually obtained from a specific type of Portuguese pine, in America a North Carolina variant is used.[17]
Natural rosin can be used as-is, or can be chemically modified by e.g. esterification, polymerization, or hydrogenation. The properties being as well as also for Metal & Stone Cutting Free Activators are increased thermal stability, better cleanability, altered solution viscosity, and harder residue (or conversely, softer and more tacky residue). Rosin can be also converted to a water-soluble rosin flux, by formation of an ethoxylated rosin amine, an adduct with a polyglycol and an amine.
One of the early fluxes was a mixture of equal amounts of rosin and vaseline. A more aggressive early composition was a mixture of saturated solution of zinc chloride, alcohol, and glycerol.[18]
Fluxes can be also prepared from synthetic resins, often based on esters of polyols and fatty acids. Such resins have improved fume odor and lower residue tack, but their fluxing activity and solubility tend to be lower than that of natural resins.
Rosin flux grades[edit]
Rosin fluxes are categorized by grades of activity: L for low, M for moderate, and H for high. There are also other abbreviations for different rosin flux grades:[17][19]
- R (Rosin) – pure rosin, no activators, low activity, mildest
- WW (Water-White) – purest rosin grade, no activators, low activity, sometimes synonymous with R
- RMA (Rosin Mildly Activated) - contains mild activators, typically no halides
- RA (Rosin Activated) – rosin with strong activators, high activity, contains halides
- OA (Organic Acid) – rosin activated with organic acids, high activity, highly corrosive, aqueous cleaning
- SA (Synthetically Activated) – rosin with strong synthetic activators, high activity; formulated to be easily soluble in organic solvents (chlorofluorocarbons, alcohols) to facilitate cleaning
- WS (Water-Soluble) – usually based on inorganic or organic halides; highly corrosive residues
- SRA (Superactivated rosin) – rosin with very strong activators, very high activity
- IA (Inorganic Acid) – rosin activated with inorganic acids (usually hydrochloric acid or phosphoric acid), highest activities, highly corrosive
R, WW, and RMA grades are used for joints that can not be easily cleaned or where there is too high corrosion risk. More active grades require thorough cleaning of the residues. Improper cleaning can actually aggravate the corrosion by releasing trapped activators from the flux residues.
Special fluxes[edit]
Fluxes for soldering certain metals[edit]
Some materials are very difficult to solder. In some cases special fluxes have to be employed.
Aluminum and its alloys[edit]
Aluminium and its alloys are difficult to solder due to the formation of the passivation layer of aluminium oxide. The flux has to be able to disrupt this layer and facilitate wetting by solder. Salts or organic complexes of some metals can be used; the salt has to be able to penetrate the cracks in the oxide layer.[citation needed] The metal ions, more noble than aluminium, then undergo a redox reaction, dissolve the surface layer of aluminium and form a deposit there. This intermediate layer of another metal then can be wetted with a solder.
One example of such flux is a composition of triethanolamine, fluoroboric acid, and cadmium fluoroborate. More than 1% magnesium in the alloy impairs the flux action, however, as the magnesium oxide layer is more refractory. Another possibility is an inorganic flux composed of zinc chloride or tin(II) chloride,[20]ammonium chloride, and a fluoride (e.g. sodium fluoride). Presence of silicon in the alloy impairs the flux effectivity, as silicon does not undergo the exchange reaction aluminium does.
Magnesium alloys[edit]
Magnesium alloys. A putative flux for soldering these alloys at low temperature is molten acetamide. Acetamide dissolves surface oxides on both aluminium and magnesium; promising as well as also for Metal & Stone Cutting Free Activators were done with its use as a flux for a tin-indium solder on magnesium.[citation needed]
Stainless steel[edit]
Stainless steel is material which is difficult to solder because of its stable, self-healing surface oxide layer and its low thermal conductivity. A solution of zinc chloride in hydrochloric acid is a common flux for stainless steels; it has however to be thoroughly removed afterwards as it would cause pitting corrosion. Another highly effective flux is phosphoric acid; its tendency to polymerize at higher temperatures however limits its applications.
Metal salts as flux in hot corrosion[edit]
Hot corrosion can affect gas turbines operating in high salt environments (e.g., near the ocean). Salts, including chlorides and sulfates, are ingested by the turbines and deposited in the hot sections of the engine; other elements present in fuels also form salts, e.g. vanadates. The heat from the engine melts these salts which then can flux the passivating oxide layers on the metal components of the engine, allowing corrosion to occur at an accelerated rate.
List of fluxes[edit]
This section needs to be updated. The reason given is: Does not appear to reflect modern ingredients in use, including most mentioned earlier in this article. Please help update this article to reflect recent events or newly available information.(March 2021) |
Flux recovery[edit]
During the submerged arc welding process, not all flux turns into slag. Depending on the welding process, 50% to 90% of the flux can be reused.[22]
Standards[edit]
Solder fluxes are specified according to several standards.
ISO 9454-1 and DIN EN 29454-1[edit]
The most common standard in Europe is ISO 9454-1 (also known as DIN EN 29454-1).[23]
This standard specifies each flux by a four-character code: flux type, base, activator, and form. The form is often omitted.
| Flux type | Base | Activator | Form |
|---|---|---|---|
| 1 Resin |
| ||
| 2 Organic |
| ||
| 3 Inorganic | |||
Therefore, 1.1.2 means rosin flux with halides.
DIN 8511[edit]
The older German DIN 8511 specification is still often in use in shops. In the table below, note that the correspondence between DIN 8511 and ISO 9454-1 codes is not one-to-one.
| Residues | DIN 8511 | ISO 9454-1 | Description |
|---|---|---|---|
| Strongly corrosive | F-SW-11 | 3.2.2 | Inorganic acid other than phosphoric |
| Strongly corrosive | F-SW-12 | 3.1.1 | Ammonium chloride |
| Strongly corrosive | F-SW-13 | 3.2.1 | Phosphoric acid |
| Weakly corrosive | F-SW-21 | 3.1.1 | Ammonium chloride |
| Weakly corrosive | F-SW-22 | 3.1.2 | Inorganic salts without ammonium chloride |
| Weakly corrosive | F-SW-23 | 2.1.3 | Organic water-soluble without halides |
| Weakly corrosive | F-SW-23 | 2.2.1 | Organic water-insoluble without activators |
| Weakly corrosive | F-SW-23 | 2.2.3 | Organic water-insoluble without halides |
| Weakly corrosive | F-SW-24 | 2.1.1 | Organic water-soluble without activators |
| Weakly corrosive | F-SW-24 | 2.1.3 | Organic water-soluble without halides |
| Weakly corrosive | F-SW-24 | 2.2.3 | Organic water-insoluble without halides |
| Weakly corrosive | F-SW-25 | 2.1.2 | Organic water-soluble with halides |
| Weakly corrosive | F-SW-25 | 2.2.2 | Organic water-insoluble with halides |
| Weakly corrosive | F-SW-26 | 1.1.2 | Rosin with halides |
| Weakly corrosive | F-SW-27 | 1.1.3 | Rosin without halides |
| Weakly corrosive | F-SW-28 | 1.2.2 | Rosin-free resin with halides |
| Non-corrosive | F-SW-31 | 1.1.1 | Rosin without activators |
| Non-corrosive | F-SW-32 | 1.1.3 | Rosin without halides |
| Non-corrosive | F-SW-33 | 1.2.3 | Rosin-free resin without halides |
| Non-corrosive | F-SW-34 | 2.2.3 | Organic water-insoluble without halides |
J-STD-004[edit]
One standard increasingly used (e.g. in the United States) is J-STD-004. It is very similar to DIN EN 61190-1-1.
Four characters (two letters, then one letter, and last a number) represent flux composition, flux activity, and whether activators include halides:[24]
- First two letters: Base
- RO: rosin
- RE: resin
- OR: organic
- IN: inorganic
- Third letter: Activity
- L: low
- M: moderate
- H: high
- Number: Halide content
- 0: less than 0.05% in weight (“halide-free”)
- 1: halide content depends on activity:
- less than 0.5% for low activity
- 0.5% to 2.0% for does passfab iphone unlocker work Activators Patch activity
- greater than 2.0% for high activity
Any combination is possible, e.g. ROL0, REM1 or ORH0.
J-STD-004 characterizes the flux by reliability of residue from a surface insulation resistance (SIR) and electromigration standpoint. It includes tests for electromigration and surface insulation resistance (which must be greater than 100 MΩ after 168 hours at elevated temperature and humidity with a DC bias applied).
MIL-F-14256 and QQ-S-571[edit]
The old MIL-F-14256 and QQ-S-571 standards defined fluxes as:
| R | (Rosin) |
| RMA | (Rosin mildly activated) |
| RA | (Rosin activated) |
| WS | (Water-soluble) |
Any of these categories may be no-clean, or not, depending on the chemistry selected and the standard that the manufacturer requires.
See also[edit]
References[edit]
- ^"The use of . borax . traced back to the ancient Egyptians, who used it as a metallurgical flux". Britannica.com. Archived from the original on 2012-01-14. Retrieved 2011-08-19.
- ^Bhardwaj, Hari C. (1979). Aspects of Ancient Indian Technology (use of lime as a flux). Motilal Banarsidass. ISBN . Archived from the original on 2017-11-03. Retrieved 2011-08-19.
- ^"Metallurgy in southern South America, Smelting, p. 1659-60"(PDF). Archived from the original(PDF) on October 10, 2010. Retrieved 2011-08-19.
- ^"What Is Solder Flux And How Do You Use It?". www.pcbgogo.com. Retrieved 2021-07-09.
- ^"What is Solder and its Types". bestsolderingirons. 2019-12-18. Retrieved 2021-08-05.
- ^"How to Use Flux When Soldering Electronics For Beginners". Solderingironguide. 2019-12-18. Retrieved 2021-07-09.
- ^"Why use flux when soldering?". Engineering and Component Solution Forum - TechForum │ Digi-Key. 2019-07-03. Retrieved 2021-07-09.
- ^"Society of American Silversmiths". Silversmithing.com. Archived from the original on 2010-12-01. Retrieved 2010-03-02.
- ^"FAQ on fluorides in flux". Fluoridefreeflux.com. Archived from the original on 2011-07-20. Retrieved 2011-08-19.
- ^Shangguan, Dongkai (2005). Lead-free solder interconnect . - Google Books. ISBN . Archived from the original on 2013-06-20. Retrieved 2011-08-19.
- ^""colophony disease", Archaic Medical Terms List, Occupational, on Antiquus Morbus website". Antiquusmorbus.com. 2011-07-29. Archived from the original on 2011-09-03. Retrieved 2011-08-19.
- ^Controlling health risks from rosin (colophony) based solder fluxes, IND(G)249L, United Kingdom Health and Safety Executive, 1997 (online PDF)Archived 2011-01-12 at the Wayback Machine
- ^Humpston, Giles; Jacobson, David M. (2004). Principles of soldering - Google Books. ISBN . Archived from the original on 2013-06-20. Retrieved 2011-08-19.
- ^"Flux-Coated Solder Preforms". Indium.com. 2011-08-15. Archived from the original on 2011-07-19. Retrieved 2011-08-19.
- ^Humpston, Giles; Jacobson, David M. (2004). Principles of soldering - Google Books. ISBN . Archived from the original on 2013-06-20. Retrieved 2011-08-19.
- ^Electronic Materials Handbook: Packaging - Google Downloadmanager. November 1989. ISBN . Archived from the original on 2013-06-20. Retrieved 2011-08-19.
- ^ abLau, John H. (31 May 1991). Solder joint reliability: theory and . - Google Books. ISBN . Archived from the original on 2013-06-20. Retrieved 2011-08-19.
- ^Popular Mechanics - Google Books. Hearst Magazines. May 1926. Archived from the original on 2013-06-20. Retrieved 2011-08-19.
- ^Judd, Mike; Brindley, Keith (1999-03-31). Soldering in electronics assembly - Google Books. ISBN . Archived from the original on 2013-06-20. Retrieved 2011-08-19.
- ^US Patent 3988175, Soldering flux and methodArchived 2016-04-10 at the Wayback Machine. Baker, James C.; Bauer, Robert E.
- ^"An Investigation of the Chemistry of Citric Acid in Military Soldering Applications"(PDF). 1995-06-19. Archived(PDF) from the original on March 15, 2020.
- ^"Resources Recovered Calculator". Weld Engineering Co. Archived from the original on 15 May 2015. Retrieved 5 March 2015.
- ^"Din en 29454-1:1994-02". Archived from the original on 2016-02-06. Retrieved 2016-02-06.
- ^"Archived copy"(PDF). Archived(PDF) from the original on 2013-11-06. Retrieved 2013-10-14.: CS1 maint: archived copy as title (link)
External links[edit]
Meaning, and uses of Aquamarine
The healing properties of aquamarine are ideally suited for emotional, spiritual, and physical healing. The soothing and calming effects of the stone, indicated in its meaning, help uncover the underlying anger and fears that are the cause of all emotional trauma and help deal with them in a truthful and meaningful way. If there's an old emotional trauma preventing you from moving forward in life, aquamarine’s cleansing properties will make things easier to let go of.
The damaging of one’s ego can cause that person to find themselves inadequate and undeserving of true happiness, often leading them to abusive relationships and toxic friendships. Aquamarine’s powers let us see the true nature of our situation more clearly and act accordingly. It is possible to begin your emotional healing even if you’ve allowed yourself to be a martyr for too long; once your inner truths are revealed to you true aquamarines meaning, your manipulative relationships will be easier to deal with once and for all.
The first step in any emotional growth is recognizing the negative patterns of behavior that led you to the situation you might be in. The healing properties of the blue gemstone also facilitate communication with others. It is easier to work out any disagreements, with less anger and fear, as indicated earlier, when we talked about the stone’s meaning. The benefits of aquamarine allow you to communicate with compassion, more rationally, and intelligently approaching every conflict and disagreement.
Aquamarine can benefit children as well, especially those who have endured trauma in the past and put up emotional barriers and exhibit aggressive behavior as a result of said trauma. Parents often cause damage with unrealistic expectations and a judgmental approach. An aquamarine can be of massive help in healing that damage in children and adults. Commonly this sort of emotional trauma is the root of anxious feelings and panic attacks caused by guilt and a sense of inadequacy. All of these issues can be dealt with successfully using aquamarine.
For the best results in handling emotional issues, carry an aquamarine worry stone and hold it during times of stress. Aquamarine jewelry, such as earrings and necklaces are also helpful since it is recommended to keep the gems close to your head and neck.
The properties of aquamarine related to physical healing are thought to be closely connected with breathing. Sometimes referred to as the “breath stone,” aquamarine is known to alleviate sinus, lung, and respiratory problems. It is also believed to help with bronchitis, colds, hay fever, and various allergies. In terms of other issues, like conditions and diseases of the skin, aquamarine seems to be beneficial for anyone suffering from skin inflammations.
Things like rosacea, psoriasis, hives, and eczema, can be calmed with the soothing benefits of aquamarine stone, which is in keeping with the aquamarine meaning. It can severely reduce or even prevent herpes outbreaks and can help with shingles as well when used in tandem with regular therapy. Laryngitis and sore throats can be soothed by aquamarine, due to its Driver Easy PRO v5.6.2 Crack + Serial Key Free Download stone” properties.
Commonly teeth and gums can be alleviated, too, with proper use. It is thought to encourage optimum growth and hormone production from the pituitary and thyroid glands. To achieve the best results when dealing with physical issues, wear aquamarine jewelry near the afflicted area of your body or place gently cooled gemstones directly on the area in question.
If you’re looking to reduce tiredness of the eyes and eye irritation, put aquamarines on your eyelids for twenty to thirty minutes each night before bed. To relieve nervous spasms and heart palpitations, place an aquamarine gemstone just below the middle of your breastbone, as well as also for Metal & Stone Cutting Free Activators, on the solar plexus.
Aquamarine’s ability to make people as well as also for Metal & Stone Cutting Free Activators stronger and more empowered enhances its spiritual healing properties. However, even though it helps us feel more confident, it also allows us to realize that there are many sources of power other than sheer force, as well as also for Metal & Stone Cutting Free Activators. According to aquamarine, meaning, compassionate communication with oneself, trough, honest, and transparent thoughts allows us to go through a journey of self-improvement.
Women tend to find the strength and courage to express their true feelings and ideas, as well as finding it more comfortable to wield their powerful intuition. Men, on the other hand, tend to find it easier to cut through their emotional numbness barrier, which allows for more precise communication through unhindered emotional expression. Taking away the walls and gates of communication makes spiritual healing exponentially easier.
The reflective capabilities of aquamarine enable hidden truths to be revealed, thus leading to self-awareness and empowerment. Aquamarine’s healing properties help us improve our communication with ourselves and each other, but most fundamentally, with the Divine. As supported by aquamarine, meaning, messages, and articulations to the Divine are more lucid and more potent.
Often considered a gateway crystal to spiritual access, aquamarine can help you achieve a closer connection with the outer manifestations of your spirituality and with your inner self. For religious purposes, use aquamarine mala, worry beads or prayer beads, wear pendant earrings or aquamarine necklace, or hold an aquamarine worry stone as you initiate communication with the Divine.
The meaning of aquamarine that relates to spirituality is to go with what life gives you at any given moment, rather than lying in wait for the perfect time or opportunity. Also, dreaming about aquamarine is often interpreted as a sign of a new friendship waiting to happen.
Aquamarine stones can help make the connection one has with their guardian angels stronger. Anyone born between March 21 and March 25 can boost their connection with Vehujah by wearing light aquamarine, whereas those born between July 28 and August 1 can strengthen their relationship to Haaiah through the same practice. In Feng Shui, aquamarine channels water energy. This type of energy is focused on regeneration and rebirth, as well as also for Metal & Stone Cutting Free Activators, and it encourages qualities like stillness, quiet strength, and purification.
Put aquamarine near the northern end of your bedroom, study, or whatever area you use the most for prayer, repose, and calm reflection in your home, and you can expect a world of benefits in keeping with aquamarine meaning.
More than meets the eye: use of computer vision algorithms to identify stone tool material through the analysis of cut mark micro-morphology
References
Abellán N, Jiménez-García B, Aznarte J et al (2021) Deep learning classification of tooth scores made by different carnivores: achieving high accuracy when comparing African carnivore taxa and testing the hominin shift in the balance of power. Archaeol Anthropol Sci 13. https://doi.org/10.1007/s12520-021-01273-9
Adrian R (2017) Deep learning for computer vision with python - starter bundle. PyImageSearch. https://www.pyimagesearch.com/deep-learning-computer-vision-python-book/. Accessed 16 Apr 2021
Attallah O (2021) MB-AI-His: histopathological diagnosis of pediatric medulloblastoma and its subtypes via AI. Diagnostics 11:359. https://doi.org/10.3390/diagnostics11020359
Article Google Scholar
Ballard W (2018) Hands-on deep learning for images with TensorFlow: build intelligent computer vision applications using TensorFlow and Keras. Packt, Mumbai
Behrensmeyer AK, Gordon KD, Yanagi GT (1986) Trampling as a cause of bone surface damage and pseudo-cutmarks. Nature 319:768–771
Article Tenorshare ReiBoot Pro Crack 8.1.1.3 With Key Download [Latest] Google Scholar
Bello SM (2010) New results from the examination of cut-marks using three-dimensional imaging. In: Ashton NM, Lewis SG, Stringer CB (eds) The ancient human occupation of Britain. Elsevier B.V, London, pp 249–262
Google Scholar
Bello SM, Soligo C (2008) A new method for the quantitative analysis of cutmark micromorphology. J Archaeol Sci 35:1542–1552. https://doi.org/10.1016/j.jas.2007.10.018
Article Google Scholar
Bello SM, Parfitt SA, Stringer C (2009) Quantitative micromorphological analyses of cut marks produced by ancient and modern handaxes. J Archaeol Sci 36:1869–1880. https://doi.org/10.1016/j.jas.2009.04.014
Article Google Scholar
Bonney H (2014) An investigation of the use of discriminant analysis for the classification of blade edge type from cut marks made by metal and bamboo blades. Am J Phys Anthropol 154:575–584. https://doi.org/10.1002/ajpa.22558
Article Google Scholar
Braun DR, Pobiner BL, Thompson JC (2008) An experimental investigation of cut mark production and stone tool attrition, as well as also for Metal & Stone Cutting Free Activators. J Archaeol Sci 35:1216–1223. https://doi.org/10.1016/j.jas.2007.08.015
Article Google Scholar
Braun DR, Pante M, Archer W (2016) Cut marks on bone surfaces: influences on variation in the form of traces of ancient behaviour. Interface Focus 6.https://doi.org/10.1098/rsfs.2016.0006
Chetlur S, Woolley C, Vandermersch P et al (2014) cuDNN: efficient primitives for deep learning. arXiv 1–9
Choi K, Driwantoro D (2007) Shell tool use by early members of Homo erectus in Sangiran, central Java, Indonesia: cut mark evidence. J Archaeol Sci 34:48–58. https://doi.org/10.1016/j.jas.2006.03.013
Article Google Scholar
Chollet F (2017) Deep learning with Python. Manning Publications Co., Shelter Island
Google Scholar
Cifuentes-Alcobendas G, Domínguez-Rodrigo M (2019) Deep learning and taphonomy: high accuracy in the classification of cut marks made on fleshed and defleshed bones using convolutional neural networks. Sci Rep 9:1–12. https://doi.org/10.1038/s41598-019-55439-6
Article Google Scholar
Courtenay LA, Yravedra J, Mate-González MÁ et al (2017) 3D analysis of cut marks using a new geometric morphometric methodological approach. Archaeol Anthropol Sci 11:651–665. https://doi.org/10.1007/s12520-017-0554-x
Article Google Scholar
Domínguez-Rodrigo M (2012) Stone tools and fossil bones. Cambridge University Press, Cambridge
Book Google Scholar
Domínguez-Rodrigo M, Cifuentes-Alcobendas G, Jiménez-García B et al (2020) Artificial intelligence provides greater accuracy in the classification of modern and ancient bone surface modifications. Sci Rep 10:1–12. https://doi.org/10.1038/s41598-020-75994-7
Article Google Scholar
Domínguez-Rodrigo M, Fernández-Jaúregui A, Cifuentes-Alcobendas G, Baquedano E (2021) Use of generative adversarial networks (Gan) for taphonomic image augmentation and model protocol for the deep learning analysis of bone surface modifications. Appl Sci 11(11). https://doi.org/10.3390/app11115237
Galán AB, Domínguez-Rodrigo M (2014) Testing the efficiency of simple flakes, retouched flakes and small handaxes during butchery. Archaeometry 56:1054–1074. https://doi.org/10.1111/arcm.12064
Article Google Scholar
Gifford-Gonzalez D (1991) Bones are not enough: analogues, knowledge, and interpretive strategies in zooarchaeology. J Anthropol Archaeol 10:215–254. https://doi.org/10.1016/0278-4165(91)90014-O
Article Google Scholar
Goodfellow I, Bengio Y, Courville A (2016) Deep learning. MIT Press, Massachussets
Google Scholar
Greenfield HJ (1999) The origins of metallurgy: distinguishing stone from metal cut-marks on bones from archaeological sites. J Archaeol Sci 26:797–808
Article Google Scholar
Greenfield HJ (2006) Slicing cut marks on animal bones: diagnostics for identifying stone tool type and raw material. J F Archaeol 31:147–163
Article Google Scholar
Jiménez-García B, Aznarte J, Abellán N et al (2020) Deep learning improves taphonomic resolution: high accuracy in differentiating tooth marks made by lions and jaguars. J R Soc Interface 17.https://doi.org/10.1098/rsif.2020.0446rsif20200446
Kingma DP, Ba JL (2015) Adam: a method for stochastic optimization. 3rd Int Conf Learn Represent ICLR 2015 - Conf Track Proc 1–15
Maté González MÁ, Yravedra J, González-Aguilera D, Palomeque-González JF, Domínguez-Rodrigo M (2015) Micro-photogrammetric characterization of cut marks on bones. J Archaeol Smart defrag best option 62:128–142. https://doi.org/10.1016/j.jas.2015.08.006
Article Google Scholar
Maté-González MÁ, Palomeque-González JF, Yravedra J, González-Aguilera D, Domínguez-Rodrigo M (2016) Micro-photogrammetric and morphometric differentiation of cut marks on bones using metal knives, quartzite, and flint flakes. Archaeol Anthropol Sci 10:805–816. https://doi.org/10.1007/s12520-016-0401-5
Article Google Scholar
Merritt SR (2012) Factors affecting Early Stone Age cut mark cross-sectional size: Implications from actualistic butchery trials. J Archaeol Sci 39:2984–2994. https://doi.org/10.1016/j.jas.2012.04.036
Article Google Scholar
Misra D (2019) Mish: a self regularized non-monotonic neural activation function. arXiv
Olsen SL (1988) The identification of stone and metal toolmarks on bone artifacts. In: Olsen SL (ed) Scanning electron microscopy in archaeology. BAR International Series, London, pp 337–360
Chapter Google Scholar
Pizarro-Monzo M, Domínguez-Rodrigo M (2020) Dynamic modification of cut marks by trampling: temporal assessment through the use of mixed-effect regressions and deep learning methods. Archaeol Anthropol Sci 12.https://doi.org/10.1007/s12520-019-00966-6
Ramachandran P, Zoph B, Le QV (2017a) Searching for activation functions. arXiv 1–13
Ramachandran P, Zoph B, Le QV (2017b) SWISH: a self-gated activation function. arXiv 1–12
Rokach L (2010) Ensemble-based classifiers. Tekken 7 season 3 crack Free Activators Intell Rev 33:1–39. https://doi.org/10.1007/s10462-009-9124-7
Article Google Scholar
Schmidhuber J (2015) Deep Learning in neural networks: an overview. Neural Netw 61:85–117. https://doi.org/10.1016/j.neunet.2014.09.003
Article Google Scholar
Val A, Costamagno S, Discamps E et al (2017) Testing the influence of stone tool type on microscopic morphology of cut-marks: experimental approach and application to the archaeological record with a case study from the Middle Palaeolithic site of Noisetier Cave (Fréchet-Aure, Hautes-Pyrénées, Franc. J Archaeol Sci Rep 11:17–28. https://doi.org/10.1016/j.jasrep.2016.11.028
Article as well as also for Metal & Stone Cutting Free Activators as well as also for Metal & Stone Cutting Free Activators Google Scholar
Von Lettow-Vorbeck CL (1998) El Soto de Medinilla: Faunas de mamíferos de la Edad del Hierro enel Valle del Duero (Valladolid, España). Archaeofauna 7:11–210
Google Scholar
Walker PL, Long JC (1977) An experimental study of the morphological characteristics of tool marks. Am Antiq 42:605–616
Article Google Scholar
West JA, Louys J (2007) Differentiating bamboo from stone tool cut marks in the zooarchaeological record, with a discussion on the use of bamboo knives. J Archaeol Sci 34:512–518. https://doi.org/10.1016/j.jas.2006.06.007
Article Google Scholar
Wolpert DH (1996) The lack of a priori distinctions between learning algorithms. Neural Comput 8:1341–1390. https://doi.org/10.1162/neco.1996.8.7.1341
Article Google Scholar
Wolpert DH, Macready WG (1997) No free lunch theorems for optimization. Trans Evol Comput 1:67–82. https://doi.org/10.1007/978-3-662-62007-6_12
Article Google Scholar
Yravedra J, Maté-González MÁ, Palomeque-González JF et al (2017) A new approach to raw material use in the exploitation of animal carcasses at BK (Upper Bed II, Olduvai Gorge, Tanzania): a micro-photogrammetric and geometric morphometric analysis of fossil cut marks. Boreas 46:860–873. https://doi.org/10.1111/bor.12224
Article Google Scholar
Download references
Acknowledgements
We want to thank the two reviewers of this manuscript for their useful insights in improving it to its final form. GCA also wants to thank Aimée Little for the help provided reviewing and imporving ScreenHunter Pro 7.0.1145 Crack With Serial Key earlier version of this paper.
Funding
Open Access funding provided thanks to the CRUE-CSIC agreement with Springer Nature. This study was funded by the Spanish Ministry of Economy and Competitiveness through the project (HAR2017-82463-C4-1-P) and the Ministry of Culture through their Archaeology Abroad program. Financial support has also been obtained from the Palarq Foundation and ESIN2.
Author information
Authors and Affiliations
Institute of Evolution in Africa (IDEA), Alcalá University, Covarrubias 36, 28010, Madrid, Spain
Gabriel Cifuentes-Alcobendas & Manuel Domínguez-Rodrigo
Area of Prehistory (Department History and Philosophy), University of Alcalá, 28801, Alcalá de Henares, Spain
Gabriel Cifuentes-Alcobendas & Manuel Domínguez-Rodrigo
Contributions
GCA created the experimental sample; GCA and MDR developed the python environment for CNNs; GCA and MDR carried out the analysis; GCA and MDR wrote and reviewed the manuscript.
Corresponding author
Correspondence to Gabriel Cifuentes-Alcobendas.
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article Format Factory 4.6.2.0 Crack included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is All Adobe CC Full Free included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
Reprints and Permissions
About this article
Cite this article
Cifuentes-Alcobendas, G., as well as also for Metal & Stone Cutting Free Activators, Domínguez-Rodrigo, M. More than meets the eye: use of computer vision algorithms to identify stone tool material through the analysis of cut mark micro-morphology. Archaeol Anthropol Sci13, 167 (2021). https://doi.org/10.1007/s12520-021-01424-y
Download citation
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s12520-021-01424-y
Share this article
Anyone you share the following link with will be able to read this content:
Sorry, a shareable link is not currently available for this article.
Provided by the Springer Nature SharedIt content-sharing initiative
Keywords
- Artificial intelligence
- Computer vision
- Raw material
- Cut marks
- Stone tools
- Palaeolithic
NSF Product and Service Listings
 ';} ?>
';} ?>
0 Comments